神经退行性病
-
Figure 1|Comparison between a healthy brain and neuropathological changes in Alzheimer’s disease.
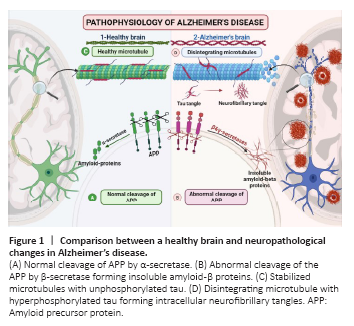
Alzheimer’s disease (AD) is a multi-etiological disorder characterized by gradual cognitive decline, and it is the most prevalent type of dementia (Panpalli Ates et al., 2016). By 2050, the number of AD patients (65 years) in the United States will more than double, from 5.8 million to 13.8 million (Zhang et al., 2021). The most common hypotheses on the etiopathogenesis of AD are the amyloid cascade hypothesis and the hyperphosphorylated tau hypothesis. Amyloid precursor protein (APP) is a widely expressed transmembrane protein in the brain. It has a remarkable physiological role during brain maturation and memory. Normally, APP is cleaved by a proteolytic enzyme called α-secretase, generating soluble peptides (Müller et al., 2017). In AD, APP undergoes proteolytic cleavage by two amyloidogenic proteases, β- and γ- secretase forming insoluble amyloid-β (Aβ40–42). These insoluble proteins clump together, forming extracellular amyloid plaques that promote immune and inflammatory responses (Lichtenthaler and Haass, 2004; Müller et al., 2017; Figure 1).
Figure 2|Graphical abstract recap the molecular mechanisms involved in the neuroprotective effects of EGFR inhibition.

Following axonal degeneration, ZNRF1 degraded AKT. As a result of Akt degradation, GSK3-β was stimulated, inhibiting collapsing response mediator protein 2 (CRMP2) that facilitated axonal growth via its interactions with the microtubules. Hence, degradation of CRMP2 resulted in the loss of axonal integrity (Wakatsuki et al., 2015). In addition, oxidative stress caused by treating neuronal cells with H2O2 or 6-hydroxydopamine or even in vivo by injury induction induced NOX in neuronal axons and activated the neuronal EGFR activity (Belambri et al., 2018). Phosphorylated EGFR activated ZNRF1 and consequently stimulated the ligase action of ZNRF1 (Araki and Wakatsuki, 2019). Interestingly, activation of the EGFR/ZNRF1/AKT/GSK3B/CRMP2 pathway in the neuronal body resulted in the stimulation of neuronal apoptosis (Cao and Fang, 2015). Accordingly, NOX is required for ZNRF1 stimulation by EGFR-dependent activation in response to axonal injury. Therefore, targeting EGFR-pluripotent oxidative stress warrants further research in neurodegenerative diseases, including AD (Figure 2c).
In 2019, Chen et al. examined the anti-inflammatory actions of EGFR inhibitor, afatinib using primary cultured astrocytes and CTX-TNA2 cells subjected to oxygen and glucose deprivation. They proved that oxygen and glucose deprivation mediated EGFR phosphorylation and stimulated downstream signaling pathways such as Akt and ERK. Furthermore, afatinib amended GFAP, cell proliferating biomarker; proliferating cell nuclear antigen levels, and hypoxia-mediated migratory capability. At the same time, afatinib suppressed cyclooxygenase-II, NO, inducible nitric oxide synthase, caspase-1, and interleukin-1β levels medium (Chen et al., 2019; Figure 2a). A recent study revealed that administration of ibrutinib, an EGFR inhibitor, ameliorated Alzheimer’s behavioral and pathological changes in transgenic mice. The neuroprotective actions were mediated via inhibition of Aβ, p-tau, cyclin-dependent kinase 5, and proinflammatory cytokines. Ibrutinib suppressed Aβ development in the primary and moderate phases of AD as demonstrated by decreased Aβ load in the hippocampus and cerebral cortex of 6-month-old transgenic AD mice (Lee et al., 2021).
CL-387,785 rescued memory decline in double transgenic mice at a low dose (5 mg/kg per day) as compared with gefitinib. The same authors used two cell cultures; HEK293-derived cell line and HEK293-derived cell line using different EGFR inhibitors such as CL-387,785 AG825, gefitinib, and lapatinib. Only CL-387,785 but not AG825, gefitinib, or lapatinib blocked the processing of APP in HEK293-derived cells but not in HEK293-derived cells (Wang et al., 2017), establishing the proof-of-principle base for the use of HER-2-targeted drugs for AD (Figure 2b). Tavassoly et al. (2021) found that administration of AZD3759, a highly selective BBB-penetrating EGFR inhibitor, for 3–10 weeks suppressed α-syncline pathology in the striatum and substantia nigra in C57BL6/C3H F1 Parkinson’s mouse model (Tavassoly et al., 2021).
Wang et al. (2013) demonstrated that the pathogenic role of EGFR is age-dependent. In 10-day-old flies expressing human pan-Aβ42, EGFR expression was elevated, and Aβ42 stimulated EGFR/PI3K, distorted the synaptic plasticity, and eventually resulted in memory impairment. Despite a decrease in total EGFR and p-EGFR in APP/PS1 8-month-old double transgenic mice, the ratio of p-EGFR/EGFR increased relative to wild-type mice (Wang et al., 2013). The previous convergent outcomes endorsed the theory that EGFR serves as a cellular receptor of amyloid peptides, as well as the Aβ-mediated stimulation of EGFR, which plays a crucial role in amyloidogenesis and memory loss. Taken together, the behavioral examination of different EGFR inhibitors warrants more studies to make us conclude that EGFR inhibition is a favorable target for slowing down Aβ-mediated memory decline (Figure 2d). TKIs studies in different AD models are summarized in Additional Table 1.