脑损伤
-
Figure 1|Silencing OLR1 attenuates functional impairment, hematoma expansion, brain water content, and neuron loss in rat brain tissues 24 hours after ICH.
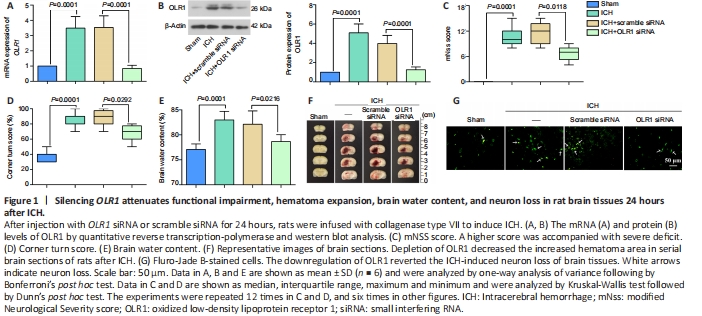
To investigate the role of OLR1 in the functional impairment after ICH, we established ICH model rats as described in Methods and evaluated the effects of OLR1 knockdown on neurological behavioral changes after ICH. As shown in Figure 1A and B, ICH-mediated induction of OLR1 mRNA and protein levels was decreased by the downregulation of OLR in brain tissues after ICH. Knockdown of OLR1 recovered the neurological dysfunction after ICH, as evidenced by the reverse of the ICH-mediated increase in mNSS score and corner turn score (Figure 1C and D). Furthermore, knockdown of OLR1 decreased the elevated brain water content and the increased hematoma area of serial brain sections of rats caused by ICH (Figure 1E and F). Moreover, downregulation of OLR1 reverted the ICH-induced neuron loss of brain tissues, as shown by Fluro-Jade B staining (Figure 1G). Together, these results indicated that decreased expression of OLR1 attenuated the functional impairment, hematoma expansion, brain water content, and neuron loss in brain tissues post-ICH.
Figure 2|Silencing of OLR1 alleviates the inflammation of rat brain tissues 24 hours after ICH.

To determine the contribution of OLR1 to inflammation, we examined the secretion of pro-inflammatory factors using ELISA. The results revealed that silencing of OLR1 reduced the increased concentration of IL-1β, IL-6, and TNF-α in the supernatant of rat brain tissues after ICH (Figure 2A–C). We next examined the infiltration of immunocytes in brain tissues by immunofluorescence staining for indicators of neutrophils, microglia/macrophages, and astrocytes (MPO, Iba1, and GFAP, respectively). As shown in Figure 2D, OLR1 knockdown reversed the elevated MPO in brain tissues induced by ICH. The same findings were observed for Iba1 and GFAP levels (Additional Figure 1). These results suggested that OLR1 knockdown alleviated the inflammation of brain tissues around hematoma after ICH.
Figure 3|Silencing of OLR1 mitigates oxidative stress in rat brain tissues after ICH.

We further investigated the effects of OLR1 on oxidative stress, which plays an important function in mediating ICH-induced injury and is characterized by the gain of ROS, MDA, 4-HNE and the loss of SOD and GSH. Dihydroethidium fluorescence assay demonstrated that the knockdown of OLR1 reversed the increased ROS in post-ICH brain tissues (Figure 3A). Furthermore, downregulation of OLR1 partially restored the reduced global SOD activity and GSH content and reversed the enhanced MDA and 4-HNE in the supernatant of rat brain tissues after ICH (Figure 3B–E). These data indicated that silencing of OLR1 mitigated oxidative stress in rat brain tissues after ICH.
Figure 4|Silencing of OLR1 suppresses ferroptosis of neurons in rat brain tissues 24 hours after ICH.

We next investigated the mechanism underlying the effects of OLR1 knockdown after ICH. GSH depletion reduces GPX4 activity and cell antioxidant capacity, resulting in the accumulation of lipid ROS and ultimately the occurrence of oxidative damage and ferroptosis (Wang et al., 2019; Li et al., 2020). Given the data on the impact of OLR1 on GSH and oxidative stress, we speculated that the loss of OLR1 might prevent secondary brain injury in ICH rats by regulating ferroptosis. We thus next examined protein expression levels of ferroptotic indicators (GPX4, FTH1, and COX-2) (Datta et al., 2013; Chen et al., 2019; Seibt et al., 2019). Western blot analysis showed that OLR1 depletion reversed the downregulation of GPX4 and FTH1 and the upregulation of COX-2 in brain tissues around hematoma after ICH (Figure 4A). Moreover, immunofluorescence staining results of GPX4 expression in NeuN-positive cells showed similar results; GPX4 was localized in cytoplasm (Figure 4B). These results indicated that silencing of OLR1 suppressed the ferroptosis of neurons in rat brain tissues induced by ICH.