视神经损伤
-
Figure 1|Two pharmacological strategies to restore rhodopsin homeostasis for treating RHO-associated retinitis pigmentosa.
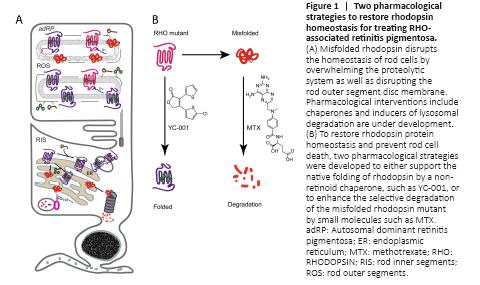
As a photopigment, rhodopsin is the most abundant protein located in the outer segments (OS) of rods, and it enables the characteristic high photosensitivity (Palczewski, 2006). Binding with an 11-cis-retinal chromophore allows photoactivation of rhodopsin and initiation of visual phototransduction. Biosynthesis of rhodopsin reaches about 6 million molecules per day per rod, lasting for the cell’s entire life span. The biosynthesis of rhodopsin requires highly coordinated regulation of the RHO gene expression, protein translation and folding, glycosylation, palmitoylation, and transport to the OS. Dysregulation of any of these steps can lead to rod stress and death. As a class A G-protein coupled receptor and a membrane protein that is not as stable as soluble proteins, the folding of a nascent apoprotein rod opsin is supported and monitored by molecular chaperones in the endoplasmic reticulum (ER) (Athanasiou et al., 2018). Misfolded rhodopsin activates the ER associated protein degradation (ERAD) pathway, leading to its degradation by the ubiquitin-proteasome system or the lysosome system (Figure 1A). Mutations that cause rhodopsin misfolding continuously activate the ERAD pathway, and the overwhelmed proteolytic system eventually results in rod death. In the OS disc membranes, the photobleached rhodopsin finally releases all-trans-retinal and converts to rod opsin for pigment regeneration. The rod opsin mutants, lacking any ligand to stabilize its structure during pigment regeneration, is highly vulnerable to misfolding; this misfolded rod opsin can disrupt the OS membrane structure and add to rod stress.
Chaperones stabilize the native folding of mutant rhodopsins: One strategy is to support the folding of the mutant rhodopsin by boosting the molecular chaperone activity, or by delivering small molecule chaperones (Figure 1B). While supplementation with vitamin A has shown efficacy in delaying disease progression in some RP patients, the lack of genetic diagnosis in these early studies makes the correlation of vitamin A with specific RP variants difficult. Genetic overexpression of molecular chaperones such as the 78-kDa glucose-regulated protein (GRP78/Bip) showed retinal protection in the transgenic rats expressing RHOP23H (Gorbatyuk et al., 2010), thus supporting the hypothesis that photoreceptors can be rescued by providing sufficient support during folding. Based on this notion, chemical chaperones (4-phenylbutyric acid) and retinoid analogues (11-cis-locked retinals, retinobenzaldehydes, beta-ionone, and NSC45012) have been tested in vitro with the demonstration of folding improvement of the mutant rod opsin and increased transport to the plasma membrane in mammalian cells (Athanasiou et al., 2018). However, most of these small molecule chaperones have no published records of in vivo efficacy notwithstanding a recent report of daily oral 4-phenylbutyric acid being retinal-protective in the RhoP23H/+ knock-in mouse model (Qiu et al., 2019).Figure 2|Identification of methotrexate (MTX) as a selective enhancer of misfolded rhodopsin degradation which showed retinal protection in the mouse model of RHODOPSIN (RHO) associated autosomal dominant retinitis pigmentosa (adRP).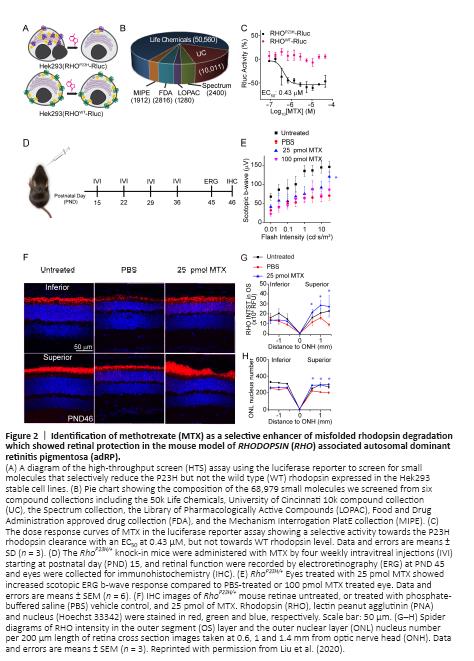
Using a luciferase reporter assay (Figure 2A), we performed a small-molecule high-throughput screening of 68,979 compounds (Figure 2B) and identified 9 compounds that selectively reduced the misfolded P23H rod opsin without an effect on the wild type rod opsin protein. Further, we found that five of these compounds, including methotrexate (MTX), promoted the degradation of misfolded rod opsin mutants. MTX, a drug commonly used off-label for treating retinal edema, already has rich documentation of its ocular pharmacokinetics and safety. MTX also showed higher selectivity in clearing mutant than WT rhodopsin (Figure 2C). Given this evidence, we selected MTX for a pilot study investigating its mechanism of action and efficacy. By pharmacologically blocking the proteasomal and the lysosomal pathways, we showed MTX induced degradation of P23H rod opsin via the lysosomal, but not the proteasomal pathway in the NIH3T3 cells. An intravitreal injection of MTX also increased the autophagy flux in vivo in the RhoP23H/+ mouse retina, suggesting MTX clears misfolded rhodopsin mainly via the autophagy-lysosome pathway. Importantly, one intravitreal injection of 25 pmol MTX at postnatal day (PND15) increased electroretinogram (ERG) response and rhodopsin level in the retinae of RhoP23H/+ knock-in mice at one month of age. Further, four weekly intravitreal injections increased the photoreceptor cell number in the retinae of RhoP23H/+ mice compared to vehicle control. Our study showed a therapeutic potential of repurposing MTX for the treatment of RHO-associated RP.