神经损伤与修复
-
Figure 1|Schematic illustration of the molecular pathways for SARS-CoV-2-induced autophagy dysregulation.
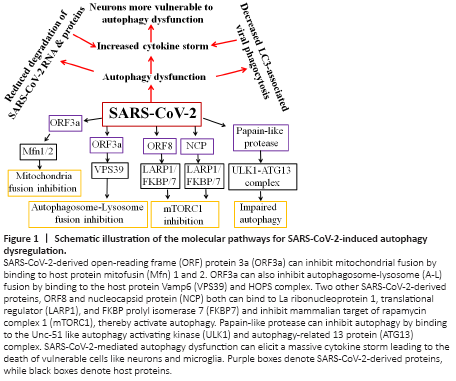
As SARS-CoV-2 enters cells through ACE2 receptors facilitated by neuropilin 1 by endocytosis, the host responds first by recognition of specific pathogen‐associated molecular patterns (PAMPs) such as spike protein and viral components by pattern recognition receptors, including the RIG‐I‐like receptors, Toll‐like receptors and inflammasomes (Li et al., 2020). Pattern recognition receptors can be localized either to cell membranes or intracellularly within various cells of the innate immune system, microglia in the brain, and many epithelial cells to facilitate pathogen recognition. SARS-CoV-2 triggered PAMPS signaling in turn induce a multitude of pathways, including pro-inflammatory responses and autophagy, Interestingly, autophagy also can regulate PAMPS and deliver exogenous viral antigens into MHC-I molecules. Moreover, autophagy can also directly impact the activation, proliferation, and differentiation of virtually all the cell types that participate in adaptive immunity including lymphocytes, antigen-presenting cells, and myeloid cells that contribute to the inflammatory response. Thus autophagy is a critical regulator of both innate and adaptive responses to viral invasion. However, SARS-CoV-2 encodes many virulence factors to evade autophagy or even turn the pathway to their own advantage. The 30 kb genome of SARS‐CoV‐2 is 100 nm in diameter and contains 14 open reading frames (ORFs) and encodes 24–27 different proteins, including the spike protein, envelope protein, membrane glycoproteins, and nucleocapsid protein (Bar-On et al., 2020). The protein ORF3a of SARS-CoV-2 dysregulates the HOPS complex by directly interacting with VPS39 thereby inhibit autophagosome-lysosome (A-L) fusion, a final ALP step critical for efficient degradation of cargo by lysosomes. Interestingly, this property of A-L fusion inhibition through HOPS-VPS39 appears to be specific to SARS-CoV-2 since very similar ORF3a of SARS-CoV was ineffective in inhibiting A-L fusion (Miao et al., 2020). Evidence also suggests that ORF3a through a 20-base sequence also inhibits mitochondrial fusion by targeting USP30 protein, a mitochondrial deubiquitinase that promotes mitochondrial fusion through deubiquitinating ubiquitylated forms of Mfn1 and Mfn2 (Singh et al., 2020). Further, SARS-CoV-2-infected cells express significantly lower levels of MTFP1 and SOCS6 which promote mitochondrial fission, suggesting that ORF3a may also compromise mitochondrial fission ultimately leading to compromise in the mitochondrial quality and early collapse of infected cells. Using affinity-purification coupled to protein identification by mass spectrometry methods, SARS-CoV-2 generated ORF8 protein, as well as nucleocapsid protein, was shown to inhibit mTORC1 by independently binding to mTORC1 pathway molecules La ribonucleoprotein 1 translational regulator and FKBP prolyl isomerase 7 (Gordon et al., 2020). mTORC1, being a negative regulator of autophagy, its SARS-CoV-2-mediated inhibition is expected to activate autophagy. However, although SARS-CoV-2-infected cells show autophagosome formation, their maturation appears to be impaired. Remarkably, a Papain-like protease of SARS-CoV-2 has been shown to directly cleave ULK1 at the recognition sequence after G499, separating its N-terminal kinase domain from a C-terminal substrate recognition region and disrupt the formation of ULK1-ATG13 complex (Mohamud et al., 2021). As ULK1 is crucial for autophagosome initiation, its cleavage is expected to stop the entire ALP as there is no autophagosome formation. Thus SARS-CoV-2 has successfully evolved several strategies to circumvent the inherently anti-viral defense capacity of autophagy (Figure 1). Other types of viruses including Coxsackievirus B3 and HIV are known to target TFEB, the master regulator of ALP, and disrupt lysosomal function. Conversely, TFEB overexpression has been shown to increase xenophagy flux and eliminate viruses. It remains to be seen whether SARS-CoV-2 proteins also directly act on TFEB and inhibit ALP. Thus, based on the available evidence SARS-CoV-2 infection in the initial 1–2 weeks may activate autophagy thereby forming more autophagosomes which aid in viral replication, but as more and more viruses are formed and RNAs and proteins get accumulated it may overwhelm the autophagy capacity after 2–5 weeks of infection thereby triggering powerful immune response resulting in cytokine storm and death in vulnerable patients.