神经退行性病
-
Figure 1|Proposed mechanisms for alterations in glucose metabolism and homeostasis in ALS.
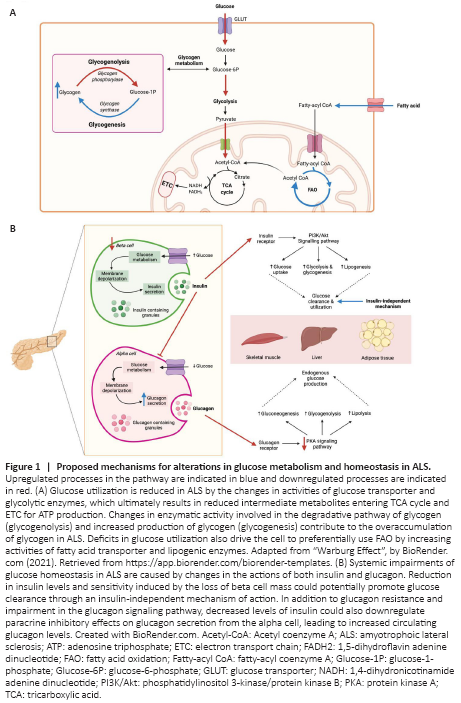
Glucose metabolism: Under normal physiological conditions, glucose metabolism begins following the uptake of extracellular glucose by the glucose transporter (GLUT) family, which is subsequently followed by a series of enzymatic reactions to generate energy in the form of adenosine triphosphate (ATP). The overall pathway of glucose metabolism is tightly regulated by both enzymes and hormones and indeed, perturbations at various steps in glucose metabolism have been described in different ALS models (Figure 1A). A reduction in glucose uptake has been identified in spinal cords and several brain regions, including motor, frontal and occipital cortex of both human and animal models of ALS, which coexist with reduced glucose consumption and functional changes in the associated regions (Tefera et al., 2021). Interestingly, glucose uptake in the peripheral tissues involved in glucose disposal, specifically skeletal muscles, adipose tissue, and liver was also observed to be increased in the SOD1G93A mice at mid-symptomatic stage of disease through a mechanism independent of insulin levels (McDonald et al., 2021). It is plausible that changes in metabolic homeostasis might underlie the upregulation of glucose uptake in an attempt to maintain glucose availability. At early pre-symptomatic stage, studies have demonstrated a selective loss of glycolytic fibers with a metabolic transition towards an oxidative metabolism in skeletal muscles of ALS. Glycolytic skeletal muscle of SOD1G93A mice showed a marked reduction in activity of phosphofructokinase, a key enzyme involves in glucose metabolism, in concomitant with increase in mitochondrial mass and expression of oxidative/intermediate myosin heavy chain (MHC) IIa isoform, as well as preservation of pure oxidative MHC I isoform at onset stage of disease (Palamiuc et al., 2015; Scaricamazza et al., 2020). This transition is likely an adaptive response to support the functional requirements by recruiting the surviving motor units in the face of defective glycolytic activity. However, despite this, the rate of ATP production, mitochondrial function and complex activities were still significantly compromised in glycolytic muscles of SOD1G93A mice, demonstrating that glucose metabolism is downregulated at the early phase of disease. Interestingly, these findings of mitochondrial defects were only detectable in the spinal cord at symptomatic stage of disease, suggesting that metabolic alterations in skeletal muscle occur independently of neuropathology and may underlie early pathological events in ALS given its role in energy homeostasis (Scaricamazza et al., 2020). Alternatively, to compensate for mitochondrial dysfunction and defective glucose utilization, transition from glucose to lipid metabolism is evident by increased dependence and capacity of lipid mobilization demonstrated in the early phase of disease in SOD1G93A mice. This metabolic shift is further exacerbated by the severe impairments in glucose oxidation as the disease progresses (Palamiuc et al., 2015; Scaricamazza et al., 2020). In addition, induced pluripotent stem cell-derived motor neurons from C9orf72-linked ALS patients and C9orf72 knockout mouse embryonic fibroblasts showed evidence of enhanced lipogenesis and lipophagy, further highlighting dysregulated lipid metabolism in ALS (Liu et al., 2018). Although utilization of fatty acids may pose an initial bioenergetic benefit, persistent fatty acid oxidation can be detrimental to mitochondrial function due to increased reactive oxygen species generated and thus potentially exacerbating degenerative processes. Taken together, whether these metabolic changes in ALS are compensatory or causative effects remain elusive and longitudinal study could potentially provide pathophysiological insights to different metabolic changes in various ALS tissues. Nevertheless, improving metabolic capacity by targeting glycolysis, providing alternative energy substrates to glucose and restoring mitochondrial biogenesis could be promising therapeutic targets for ALS.
Hormonal regulation of glucose homeostasis: Glucose metabolism is tightly regulated through the opposing effects of two hormones released from the pancreas: insulin and glucagon. In response to high blood glucose concentrations, insulin is released from pancreatic β-cells and binds to its receptors in the liver, skeletal muscle, and adipose tissue to reduce blood glucose levels. In the liver, insulin works by promoting the production of glycogen and fatty acids. In adipose tissue and skeletal muscle, insulin promotes the uptake of glucose from the blood by translocating GLUT4, an insulin-dependent transporter to the cell membrane. Indeed, both protein levels and translocation of GLUT4 were significantly downregulated in response to insulin in skeletal muscles of MLC/SOD1G93A and TDP-43 (A315T) transgenic mouse model of ALS, resulting in reduced blood glucose clearance capacity (Stallings et al., 2013; Dobrowolny et al., 2018). It would be expected that muscle atrophy would contribute to the insulin resistance that is observed in ALS; however, studies have found that this alone is unlikely to contribute to aberrant glucose handling (Pradat et al., 2010). In spite of this, insulin-independent mechanisms of glucose uptake have been identified, possibly to compensate for the insensitivity of tissues to insulin. The study performed by McDonald et al. (2021) has confirmed evidence for this, and has also identified changes to β-cells in the pancreas with disease progression. At the onset of symptoms, pancreatic β-cells remained intact and insulin secretion was unchanged, however, as disease progressed β-cell mass decreased. It is possible that the loss of β-cells in the pancreas at the later stage of disease could be due to overstimulation. Specifically, high blood glucose levels lead to the β-cells secreting insulin but as the ALS target tissues are unresponsive, this causes the blood glucose levels to remain unchanged, hence leading to more β-cell stimulation (Figure 1B). Eventually, this can cause detrimental damage to β-cells and their function, leaving the regulation of blood glucose levels to other insulin-independent mechanisms – some of which may include other GLUT isoforms present in the body. In support of this notion, overexpression of GLUT3 in the motor neurons of TDP-43 (WT) or TDP-43 (G298S) expressing drosophila has been shown to alleviate features of TDP-43 proteinopathy, possibly by increasing glucose availability and restoring metabolic homeostasis (Manzo et al., 2019). However, whether such protective effect occurs in the systemic will require further study. There are currently no published studies examining how β-cells are lost in the context of ALS, which is another possible avenue to explore in future studies.