神经损伤与修复
-
Figure 1|BRAFV600E expression leads to neuronal cell death and glial cell proliferation in primary mouse cortical mixed culture.
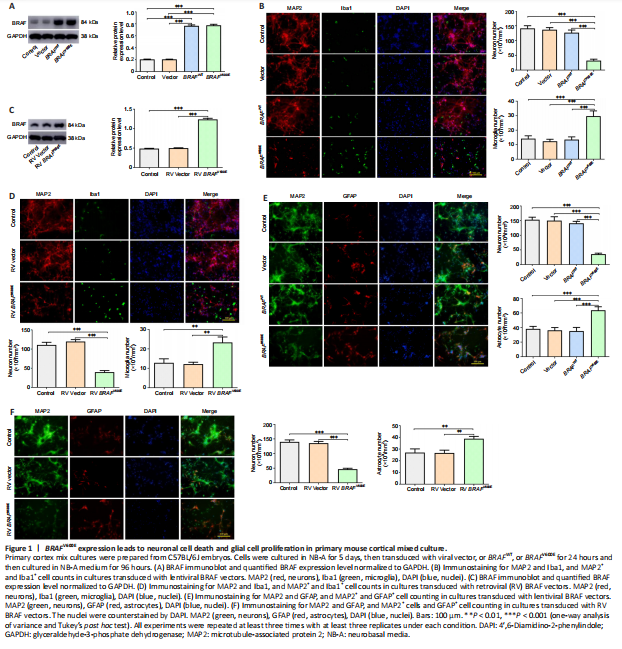
Primary cortical mixed culture consisting of neurons and glial cells (astrocytes and microglia) was transduced with LV expressing human BRAFV600E, human BRAFWT, or control vector. BRAF overexpression was detected by western blotting, which showed significantly increased BRAF in transduced cells than vector or non-transduced control cells (P < 0.001; Figure 1A). Immunostaining for MAP2, a marker of neurons, and Iba1, a marker for microglia, 120 hours after the transduction showed a significant decrease in the number of neurons and a significant increase in the number of microglia in the mixed culture transduced with BRAFV600E than vector or BRAFWT (P < 0.001; Figure 1B). Similarly, immunostaining for MAP2 and GFAP, a marker for astrocytes, showed a significant decrease in the number of neurons and a significant increase in the number of astrocytes in the culture transduced with BRAFV600E than vector or BRAFWT (P < 0.001; Figure 1E). BRAFWT did not change the number of either MAP2+ or Iba1+ or GFAP+ cells (Figure 1B and E). These results suggest that BRAFV600E expression in the cortical mixed culture may lead to different outcomes for neurons and glial cells.
The mixed cultures were transduced with BRAFV600E or control vector by RV. Unlike LV, which is capable of infecting both neurons and glial cells, RV infects only dividing cells, not postmitotic neurons. Immunoblotting showed significantly higher BRAF expression in RV transduced cells than vector or non-transduced control cells (Figure 1C). After 120 hours, MAP2 and Iba1 staining showed a significant increase in the number of microglia (P < 0.01) and a significant decrease in the number of neurons in RV BRAFV600E-transduced culture (P < 0.001) than RV vector-transduced culture (Figure 1D). BRAFV600E transduction resulted in a significant increase in the number of astrocytes (P < 0.01) and a significant decrease in the number of neurons (P < 0.001) than RV vector-transduced cells, which were demonstrated by MAP2 and GFAP staining (Figure 1F). These findings suggest that the mutant BRAF may cause neuronal loss through the proliferation of microglia and/or astrocytes.
Figure 2|BRAFV600E expression in astrocytes promotes proliferation.
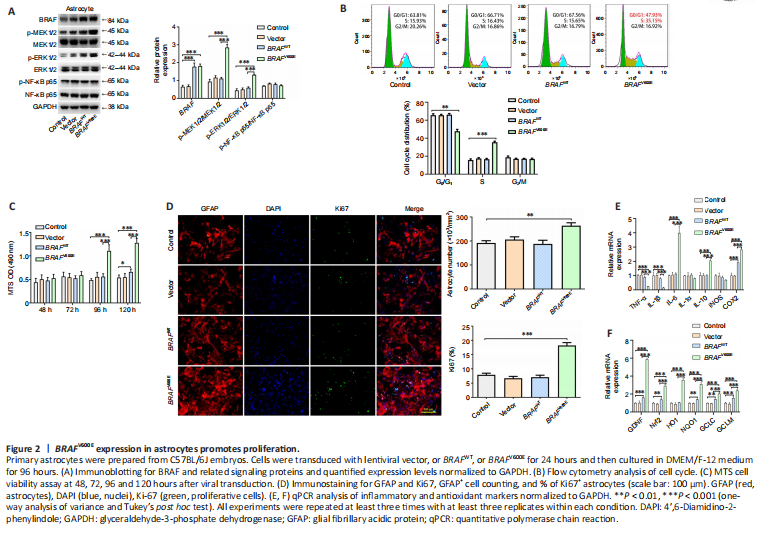
To investigate the effect of BRAFV600E on individual cell types, we next transduced primary astrocytes prepared from C57BL/6 mouse embryos with lentiviral BRAFV600E, BRAFWT, and control vector, respectively. Western blotting showed that 120 hours after the transduction, expression levels of MAP/phosphorylated MEK1/2 (p-MEK1/2), phosphorylated ERK1/2 (p-ERK1/2) in BRAFV600E -transduced astrocytes were significantly increased compared with the vector-transduced or non-transduced control groups, suggesting activation of the ERK pathway by BRAFV600E (P < 0.001; Figure 2A). Flow cytometry showed that there was a significantly lower percentage of cells in G0/G1 phase (P < 0.01) and a significantly higher percentage of cells in the S phase in BRAFV600E-transduced astrocytes as compared with vector or non-transduced control groups (P < 0.001; Figure 2B). Despite an increase in BRAF expression by nearly 3-fold, BRAFWT did not change cell cycle distribution in cultured primary astrocytes, which can be explained by the approximately 500-fold greater kinase activity of BRAFV600E than BRAFWT (Wan et al., 2004). MTS assay revealed that astrocyte viability was increased significantly 96 and 120 hours after BRAFV600E transduction compared with vector or non-transduced control group (P < 0.001; Figure 2C). Immunostaining for GFAP and Ki67, an indicator of proliferation, showed that a total number of GFAP+ cells and percentage of Ki67+ cells in the BRAFV600E -transduced group were significantly higher than vector or non-transduced control group 96 hours after transduction, while transduction with BRAFWT showed no significant difference compared with vector or the control group (Figure 2D). IL-6, IL-10, and cyclooxygenase 2 (COX2) were significantly increased at mRNA level whereas TNF-α and IL-1β levels were decreased after BRAFV600E transduction (P < 0.001; Figure 2E). In addition, nuclear factor erythroid 2 (NF-E2)-related factor 2 (Nrf2) and its downstream genes heme oxygenase 1 (HO1), quinone oxidoreductase 1 (NQO1), glutamate-cysteine ligase catalytic subunit (GCLC), and glutamate cysteine ligase modifier subunit (GCLM) were increased significantly in BRAFV600E -transduced astrocytes. In addition, there was a dramatic increase in glial-derived neurotrophic factor (GDNF) mRNA level in BRAFV600E-transduced cells (P < 0.001; Figure 2F).
Figure 3|BRAFV600E expression in microglia induces cell proliferation and activation through ERK.

We next employed mouse primary microglia to further characterize the effects of BRAFV600E on microglial cells. Cells were transduced with BRAF vectors for 120 hours. BRAFV600E transduction significantly increased p-MEK1/2 and p-ERK1/2 compared with vector and non-transduced control groups (P < 0.001). In addition, p-NF-κB p65 and p-JNK were increased (P < 0.001; Figure 3A). To determine the roles of activated ERK and JNK in BRAFV600E effects, we transduced microglial cells with siRNA targeting JNK (si-JNK), siRNA targeting ERK (si-ERK), and control-siRNA. Knockdown efficiency of si-JNK and si-ERK was demonstrated by western blotting (Figure 3B and C). After treatment with si-ERK, BRAFV600E -transduced microglia showed significantly downregulated ERK as compared with control-siRNA-treated cells expressing BRAFV600E (P < 0.001). NF-κB phosphorylation was similarly downregulated. si-JNK did not change p-NF-κB in BRAFV600E -transduced microglia (Figure 3D). Iba1 staining showed that the cell number and percentage of Ki67+ cells in the BRAFV600E -treated group were significantly increased compared with vector and non-transduced control groups (P < 0.001). The BRAFWT-treated group did not show a significant difference from vector and non-transduced control groups (Figure 3E). si-ERK treatment blocked BRAFV600E -induced proliferation of microglia. Si-JNK did not change the number of Iba1+ cells and the percentage of Ki67+ cells in BRAFV600E-transduced cells (Figure 3E). Non-transduced control microglia displayed the typical branching shape at the resting state. Transduction with vector, BRAFWT, and BRAFV600E plus si-ERK caused no obvious morphological changes compared with the control group. BRAFV600E and BRAFV600E plus si-JNK transduction induced enlargement of the microglial cell body, loss of ramifications, and development of an amoeboid shape (Figure 3E and F).
Flow cytometry showed that, compared with the vector group, the percentage of S-phase microglia in the BRAFV600E-treated group was increased significantly (P < 0.001; Figure 3G), and this effect of BRAFV600E was diminished when cells were treated with si-ERK but not si-JNK (Figure 3H). MTS assay revealed significantly higher cell viability after transduction with BRAFV600E for 96 hours compared with vector (P < 0.05) and non-transduced control groups (P < 0.01), the change becomes more significant at 120 hours (P < 0.001), and si-ERK reversed this effect (Figure 3I). Inflammatory markers TNF-α, IL-1α, IL-1β, IL-6, human inducible NO synthase, and COX2 were significantly increased at the mRNA level after transduction with BRAFV600E for 120 hours (P < 0.001). BRAFWT-transduced microglia displayed similar trends but to a lesser extent (P < 0.001), supporting BRAF activity dependence on the changes. si-ERK significantly inhibited BRAFV600E-induced inflammatory gene expression (P < 0.001), while si-JNK did not reverse the effect (Figure 3K). ELISA and nitrite release showed that inflammatory factors (IL-1β, IL-6, and TNF-α) and NO secreted by microglia after treatment with BRAFV600E were significantly higher than vector and non-transduced control groups (P < 0.001). si-ERK, but not si-JNK, reversed the secretion of inflammatory factors (IL-1β, IL-6, and TNF-α) and NO-induced by BRAFV600E (P < 0.001; Figure 3J and L).
Figure 4|Conditioned medium from BRAFV600E-expressing microglial cells but not astrocytes induces neuronal cell death.

We next treated primary cortical neurons with conditioned medium from astrocytes transduced with BRAFV600E, BRAFWT, vector, or from non-transduced control cells for up to 72 hours. Astrocytes transduced with BRAFV600E were proliferating with upregulated GDNF, certain cytokines, and antioxidants as shown in Figure 2F. However, no significant difference in the number of MAP2+ was demonstrated in all astrocyte-conditioned medium-treated groups compared with the control group (Figure 4A and B). LDH release showed no significant change among groups at 24, 28, and 72 hours (Figure 4C).
Primary cortical neurons were in parallel treated with conditioned medium from primary microglia transduced with BRAFV600E, BRAFWT, vector, or from non-transduced control cells for 72 hours. A significant decrease in the number of MAP2+ cells was demonstrated in cells treated with microglia-BRAFV600E conditioned medium (P < 0.001; Figure 4F). BRAFV600E conditioned medium also induced LDH release in primary neurons (Figure 4G and H). Treatment with microglia-control, microglia-vector, and microglia-BRAFWT conditioned medium did not result in changes compared to the control group (Figure 4D and F). Similar neurotoxicity was demonstrated in the microglia-BRAFV600E + si-JNK conditioned medium group, but not in the microglia- BRAFV600E + si-ERK conditioned medium group (Figure 4D, E, and H), suggesting that the indirect, toxic effects of BRAFV600E on neurons resulted from ERK-mediated changes in microglia.
Figure 5|BRAFV600E expression in neurons promotes cell death through the JNK pathway.
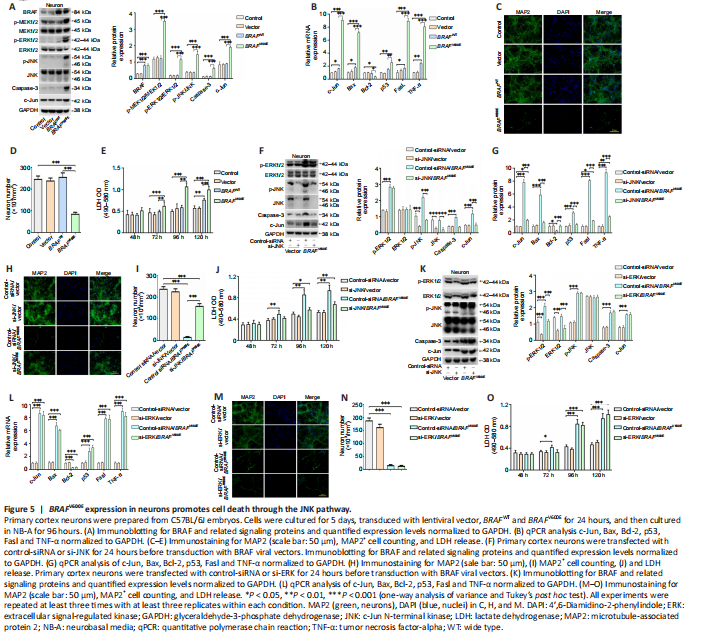
To characterize direct neuronal effects of BRAFV600E, primary cortical neurons were transduced with BRAFV600E BRAFWT, and vector for 24 hours. The cells were then cultured for 96 hours in neural culture basal medium. Western blotting confirmed BRAF overexpression and significant higher levels of p-MEK1/2, p-ERK1/2, p-JNK, caspase-3 and c-Jun in the BRAFV600E-treated group as compared with vector and non-transduced control groups (P < 0.001; Figure 5A). Marked increases in c-Jun, p53, Fas-ligand (FasL), TNF-α, and B cell leukemia/lymphoma-2 (Bcl-2) associated X protein (Bax) at the mRNA level were observed after transduction with BRAFV600E for 120 hours. Bcl-2 was significantly decreased following BRAFV600E transduction (P < 0.05; Figure 5B). BRAFV600E induced significant neurotoxicity as demonstrated by loss of MAP2+ cells and release of LDH in BRAFV600E-transduced neurons compared with vector- and non-transduced neurons (P < 0.001; Figure 5C–E).
si-JNK and si-ERK were used to block signals downstream to BRAF. Knockdown efficiency of si-JNK and si-ERK was verified by western blotting (Additional Figure 3). Significantly reduced p-JNK, caspase-3, and c-Jun were demonstrated in neurons treated with si-JNK + BRAFV600E as compared with cells treated with BRAFV600E (P < 0.001; Figure 5F). At mRNA level, si-JNK reversed BRAFV600E-induced changes in c-Jun, Bax, Bcl-2, p53, Fasl and TNF-α in cultured cortical neurons (Figure 5G). Treatment with si-ERK blocked p-ERK and total ERK expression induced by BRAFV600E (Figure 5K). There was no significant difference in protein expression of p-JNK, caspase-3 and c-Jun or mRNA expression of c-Jun, Bax, Bcl-2, p53, Fasl and TNF-α between BRAFV600E and BRAFV600E + si-ERK groups (Figure 5H and L). These results suggest that activation of the JNK pathway by BRAFV600E is independent of BRAFV600E-induced ERK activation. MAP2 staining as well as LDH assay showed significantly reduced number of MAP2+ cells and higher LDH release in cells transduced with BRAFV600E (P < 0.001; Figure 5C–E) and the changes were partially reversed after co-treatment with si-JNK (P < 0.01; Figure 5H–J). si-JNK also inhibited JNK in vector-transduced neurons but without significant influences on c-Jun, Bax, Bcl-2, p53, Fasl and TNF-α and neuronal survival (Figure 5F and G), which suggests that a threshold JNK activity may be required in activating downstream genes and inducing neuronal cell death in this culture system (Hollville et al., 2019). Co-treatment with si-ERK did not change MAP2+ cell number and LDH release in BRAFV600E-transduced cells (Figure 5N and O). Despite reported role of ERK in neuronal survival (Hossain et al., 2013) and a trend for reduced number of MAP2+ cells and increased LDH release at 120 hours, there was no statistically significant difference between si-ERK and control-siRNA treated vector-transduced neurons at the time points investigated (Figure 5M).