脊髓损伤
-
Figure 1|Ruxolitinib enhances motor function recovery in a mouse model of SCI.

To investigate the potential therapeutic effects of RUX on functional recovery after SCI, we established an experimental mouse model to simulate SCI. We administered RUX to both the SCI group and the Sham group at 1, 7, 14, and 28 days post-injury and monitored the body weight. Although the body weight in the SCI group decreased compared with that in the Sham group, there was no significant difference in body weight between the vehicle-treated and RUX-treated groups (Figure 1B), indicating no adverse effects of RUX treatment on the mice.
Motor function and axon regeneration were assessed through footprint analysis, swimming score, Basso mouse scale scoring, and histological staining (hematoxylin and eosin and immunofluorescence staining). In the footprint analysis, which assessed hindlimb function post-SCI, RUX treatment improved the hindlimb function of SCI mice. Mice in the SCI + RUX group had longer stride lengths and wider step widths than those in the SCI + vehicle group (Figure 1C–E). Similarly, in the swimming score test, mice treated with RUX showed significantly improved motor function (Figure 1F and G). To assess motor function recovery from day 1 to day 28 post-injury, we used the widely accepted Basso mouse scale score. Although the Sham group maintained scores within the normal range, both SCI groups exhibited decreased scores after SCI. From day 7 to day 28, the SCI + RUX group had significantly higher scores compared with the SCI + vehicle group, indicating that RUX treatment effectively improved motor recovery (Figure 1H). Compared with vehicle treatment, RUX treatment significantly reduced the area of spinal cord damage (Figure 1I and J). To examine the effects of RUX treatment on neuronal survival after SCI, immunofluorescence staining was performed using the neuronal marker NeuN within a defined distance from the injury edge (1000 μm). We found a significantly higher number of surviving NeuN+ neurons in the SCI + RUX group compared with the SCI + vehicle group (Figure 1K and L). Together these results indicated that RUX promotes motor function recovery and neuronal survival after SCI.
Figure 3|RUX restores the expression of EAAT2 after spinal cord injury.

Our results suggested that RUX may exert therapeutic effects against SCI through a mechanism involving upregulation of EAAT2. Previous studies reported that EAAT2 is remarkably downregulated after SCI. The reduced expression of EAAT2 in astrocytes disrupts glutamate homeostasis in damaged spinal cord tissue, leading to severe excitotoxicity in neurons (Lepore et al., 2011a; Alijanpour et al., 2023). To analyze the dynamic expression profile of EAAT2 after SCI, we assessed EAAT2 expression on days 0, 3, 7, 14, and 28 after SCI using western blotting and qPCR. Western blotting results showed that EAAT2 expression decreased after injury, with the lowest expression on day 7 (Figure 3A and B). Similar results were observed using qPCR (Figure 3C). We next examined whether RUX influenced the expression of EAAT2 after SCI. Treatment with RUX effectively counteracted the downregulation of EAAT2 at both protein and mRNA levels (Figure 3D–F), which is consistent with the results of transcriptomic sequencing. To further examine the effect of RUX on EAAT2 downregulation, immunofluorescence staining was performed on damaged spinal cord tissue samples from the Sham, SCI + vehicle, and SCI + RUX groups on day 7 post-injury. The lesion center was characterized by fibrotic scarring and the presence of macrophage-associated glial elements. Astrocytes surrounding the glial scar were designated as scar-forming reactive astrocytes, with further subclasses including reactive astrocytes and na?ve astrocytes (Li et al., 2023b). The fluorescence intensity of EAAT2 in the reactive astrocyte region surrounding the injury site was significantly decreased in the SCI + vehicle group compared with the sham group. In contrast, the SCI + RUX group exhibited a substantial increase in EAAT2 fluorescence intensity around GFAP+ cells (Figure 3G and H).
Glutamate levels were particularly high in damaged spinal cord tissues. Treatment with RUX effectively reduced glutamate levels, indicating that RUX may restore glutamate homeostasis and alleviate excitotoxicity induced by excess glutamate (Figure 3I). As a potent inhibitor of the JAK–STAT pathway, RUX effectively suppresses STAT3 phosphorylation in both in vitro and in vivo settings. STAT3 plays an important role in astrocyte activation, and its downregulation restores the glutamate-clearing capacity of astrocytes and attenuates methamphetamine withdrawal responses in mice (Shi et al., 2021). SCI is associated with JAK2-STAT3 pathway activation, manifested by elevated levels of phosphorylated JAK2 and STAT3. We found that RUX inhibits the phosphorylation of both JAK2 and STAT3 (Figure 3J–L), so we speculate that its therapeutic effect involves restoring EAAT2 expression by inhibiting STAT3 activation.. In summary, these results suggest that RUX reverses the downregulation of EAAT2 after SCI, thereby restoring glutamate homeostasis and reducing excitotoxicity induced by excess glutamate. Additionally, these results highlight the potential involvement of activated STAT3 in the protective mechanism of action of RUX.
Figure 4|RUX inhibits the activation and inflammatory responses of neurotoxic astrocytes after spinal cord injury in vivo.

After SCI, astrocytes undergo activation, exhibit heightened reactivity, and lose their original cellular morphology and some functions during phenotypic transformation. Additionally, they secrete inflammatory cytokines, acquiring neurotoxic characteristics (Fan et al., 2016; Lee et al., 2018; Zeng et al., 2019; Bretheau et al., 2022). Therefore, we investigated the influence of RUX on astrocyte activation and the secretion of pro-inflammatory cytokines after SCI. Immunofluorescence staining of injured spinal cord tissues on day 7 revealed that astrocytes in the SCI + vehicle group were significantly activated, exhibiting pronounced hypertrophy and transformation into neurotoxic astrocytes. Additionally, the injury site had abundant C3+ cells. In contrast, in the SCI + RUX group, astrocyte hypertrophy was considerably mitigated, resulting in a significant decrease in the fluorescence intensity of C3+ astrocytes at the injury site (Figure 4A and B). Consistently, western blotting showed that C3 expression substantially increased after SCI but decreased after treatment with RUX (Figure 4C and D). These results suggest that RUX effectively inhibits the activation of neurotoxic astrocytes after SCI.
Microglia/macrophages exist on a spectrum from M1 to M2, characterized by the expression of iNOS and Arg1 markers, respectively. After SCI, M1 microglia/macrophages aggravate inflammation and neural cell damage, whereas M2 microglia/macrophages alleviate inflammation, neural cell protection, and repair (Fan et al., 2018, 2020). To investigate whether RUX influences M1 polarization of microglia/macrophages, immunofluorescence staining was performed on injured spinal cord tissues on day 7 after SCI. The results revealed an increase in the population of M1 microglia/macrophages in the SCI + vehicle group, and a marked reduction in M1 microglia/macrophages numbers in the SCI + RUX group, with no significant alterations in the expression of IBA1, a microglial/macrophage marker, between the two groups (Additional Figure 2). These findings imply that RUX suppresses M1 polarization and inflammatory responses of microglia/macrophages following SCI.
Similar to microglia/macrophages, astrocytes activate the nuclear factor kappa-B (NFκB) signaling pathway after SCI, triggering the secretion of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α (Zhang et al., 2022). Western blotting revealed that treatment with RUX suppressed the phosphorylation of NFκB after SCI (Figure 4E and F). Immunofluorescence staining revealed a significant reduction in the expression of IL-1β, IL-6, and TNF-α in astrocytes upon RUX treatment (Figure 4G–J). To further substantiate the potential of RUX to diminish pro-inflammatory cytokine secretion in vivo, enzyme-linked immunosorbent assay was used to measure the relative concentrations of IL-1β, IL-6, and TNF-α in spinal cord tissues from each group. The concentrations of the three pro-inflammatory cytokines were significantly increased after SCI, with IL-6 exhibiting the most substantial increase. However, treatment with RUX decreased the levels of these pro-inflammatory cytokines (Figure 4K), a trend further confirmed by qPCR results (Figure 4L–N). These findings collectively emphasize the remarkable ability of RUX to effectively suppress the activation of neurotoxic astrocytes and reduce the levels of pro-inflammatory cytokines associated with the NFκB signaling pathway following SCI.
Figure 5|RUX rescues astrocyte EAAT2 loss and restores glutamate uptake in vitro.
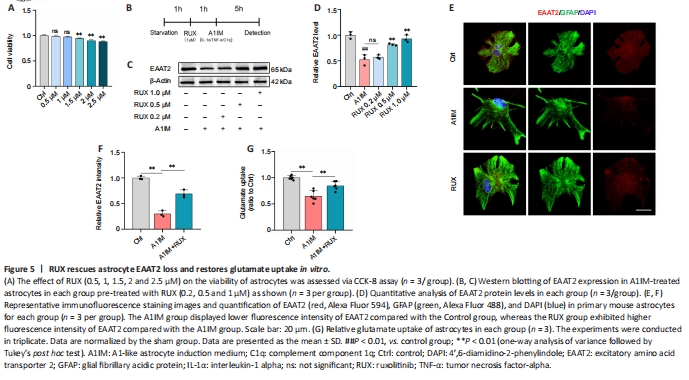
To determine the optimal concentration of RUX for intervention in an in vitro cellular model, CCK-8 assay was used to assess the proliferation and viability of astrocytes treated with RUX. Astrocytes were exposed to either vehicle (1% DMSO) or various concentrations of RUX (0.5, 1, 1.5, 2, and 2.5 μM) for 24 hours, and CCK-8 assays were performed. We observed that 0.5 and 1 μM RUX displayed no significant cytotoxicity, whereas 1.5, 2, and 2.5 μM RUX significantly affected astrocyte proliferation and viability, indicating notable cytotoxicity (Figure 5A). Therefore, RUX was used at a concentration of < 1 μM for subsequent in vitro experiments.
In vivo experiments demonstrated that RUX exerted therapeutic effects against SCI by restoring EAAT2 expression in astrocytes. To validate these effects in vitro, astrocytes were treated with either 1% DMSO or RUX at different concentrations (0.2, 0.5, and 1 μM) for 1 hour, followed by treatment with A1IM (IL-1α, TNF-α, and C1q) or PBS for 5 hours. EAAT2 expression was measured using western blotting and immunofluorescence staining (Figure 5B). A1IM treatment significantly reduced EAAT2 expression in astrocytes, and 0.2 μM RUX led to an increase in EAAT2 expression compared with A1IM treatment; however, the difference was not statistically significant. As the concentration of RUX increased, the expression of EAAT2 increased significantly (Figure 5C and D). These results indicated that RUX rescued the A1IM-induced downregulation of EAAT2 in astrocytes. The immunofluorescence staining results were in line with the findings from western blotting (Figure 5E and F).
We next assessed the glutamate uptake capacity of astrocytes. Following A1IM treatment, astrocytes were treated with 100 nM glutamate. A1IM treatment led to a marked decrease in EAAT2 expression, resulting in a substantial reduction in glutamate uptake capacity. However, pre-treatment with RUX significantly enhanced the glutamate uptake capacity of astrocytes (Figure 5G). These findings suggest that RUX restored EAAT2 expression and glutamate uptake induced by IL-1α, TNF-α, and C1q in astrocytes, aligning with the outcomes of in vivo experiments.
Figure 6|RUX restores EAAT2 loss in astrocytes by inhibiting the activation of STAT3 in vitro.
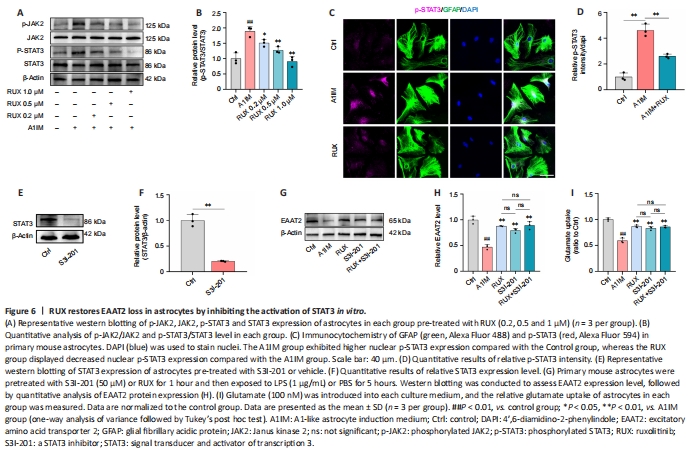
To investigate whether STAT3 was involved in RUX-mediated restoration of EAAT2 expression, astrocytes were treated with either 1% DMSO or RUX (0.2, 0.5, and 1 μM) for 1 hour, followed by treatment with A1IM (IL-1α, TNF-α and C1q) or PBS for 5 hours. JAK2 and STAT3 activation was assessed using western blotting and immunofluorescence staining. A1IM treatment activated JAK2 and STAT3, whereas RUX suppressed JAK2 and STAT3 activation in a concentration-dependent manner (Figure 6A and B). To further evaluate whether RUX influenced nuclear phosphorylated STAT3 levels in primary astrocytes stimulated by A1IM, mouse primary astrocytes were treated with control (1% DMSO) or RUX (1 μM) for 1 hour, followed by A1IM or PBS treatment for 5 hours. Immunofluorescence staining revealed that A1IM significantly increased nuclear p-STAT3 levels compared with control treatment (Figure 6C and D). In contrast, RUX treatment significantly reduced nuclear p-STAT3 levels in A1IM-stimulated mouse primary astrocytes (Figure 6C and D). These results indicated that RUX inhibited STAT3 activation in astrocytes treated with A1IM.
The STAT3 inhibitor S3I-201 (50 μM) was used to inhibit STAT3 expression, either independently or in combination with RUX pre-treatment. Astrocytes were then treated with A1IM to induce reactivity. Western blotting revealed that S3I-201 significantly inhibited STAT3 expression (Figure 6E and F). Compared with RUX or S3I-201 treatment, combination treatment with S3I-201 and RUX (S3I-201 + RUX group) did not restore EAAT2 expression or enhance glutamate uptake in A1IM-stimulated astrocytes (Figure 6G–I). These results indicate that RUX restores EAAT2 expression and glutamate uptake in astrocytes by inhibiting STAT3 activation.
Figure 7|RUX alleviates GLU-induced neuronal excitotoxicity by enhancing the GLU-clearing capacity of astrocytes.

We initially monitored calcium influx in neurons using Fluo-4 AM calcium imaging, as glutamate activation of neurons can significantly increase intracellular calcium. The astrocytes significantly reduced glutamate levels in the culture medium, resulting in a substantial reduction of Fluo-4 signal (?F/F0) in neurons (Figure 7A; instantaneous calcium imaging at 180 seconds, Figure 7B). The statistical analysis of Fluo-4 signals at 180 seconds is shown in Figure 7C. Neurons in the glutamate group exhibited a significant increase in calcium influx. Notably, A1IM treatment of astrocytes led to elevated glutamate levels in the medium, resulting in a substantial increase in calcium influx in neurons. Treatment with RUX restored the glutamate-clearing capability of A1IM-stimulated astrocytes, resulting in a decrease in calcium influx in neurons and the subsequent restoration of calcium homeostasis. Increased calcium influx contributes to calcium imbalance in neurons, triggering the generation of ROS in mitochondria. Flow cytometry revealed that treatment with RUX significantly reduced ROS levels in neurons, which was consistent with the calcium imaging results (Figure 7D and E).
Western blot analysis was conducted to assess the levels of pro-apoptotic proteins Bax and cleaved caspase-3 and the anti-apoptotic protein Bcl-2. In comparison with the PBS group, the glutamate group exhibited significantly elevated expression of Bax and cleaved caspase-3, along with reduced expression of Bcl-2. The AST group displayed decreased expression of Bax and cleaved caspase-3, while exhibiting increased levels of Bcl-2 when compared with the glutamate group (Figure 7F and G). Conversely, the A1IM group showed increased expression of Bax and cleaved caspase-3 and decreased Bcl-2 expression. Notably, the RUX group showed reduced expression of Bax and cleaved caspase-3 and an increase in Bcl-2 levels, demonstrating the protective effects of RUX against apoptosis.
To further prove the protective effect of RUX on neuronal apoptosis, flow cytometry was performed on cells in each group. The number of apoptotic neurons in glutamate group was significantly increased, whereas that in the AST group was significantly decreased. Additionally, apoptotic cells were significantly increased in the A1IM group compared with the AST group, and the proportion of apoptotic cells in the RUX group was significantly decreased compared with the A1IM group, indicating that A1IM could damage the neuroprotective effect of astrocytes, whereas RUX could restore the neuroprotective effect of astrocytes (Additional Figure 3).
To validate the neuroprotective effects of RUX and assess its effect on the complexity of dendritic branching, we performed immunofluorescence staining for MAP2, followed by Sholl analysis. This analysis provides insights into neuronal complexity by revealing the number of intersections at different distances from the cell body. Compared with the PBS group, the glutamate group exhibited significantly reduced dendritic branching complexity. Treatment with A1IM aggravated synaptic damage, whereas treatment with RUX restored the complexity of dendritic branching (Figure 7H and I).
Figure 8|RUX inhibits activation and inflammatory responses of neurotoxic astrocytes in vitro.
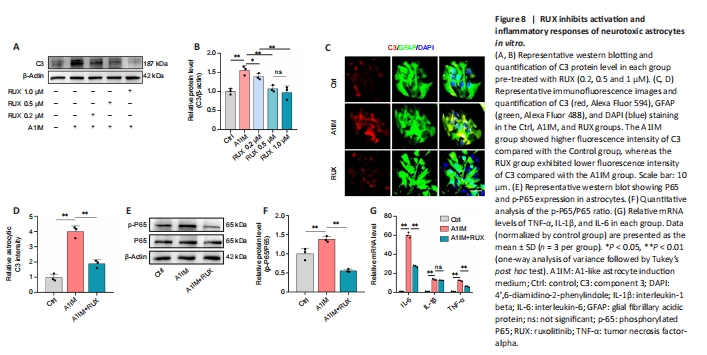
In vivo experiments demonstrated that RUX inhibited the activation and inflammatory responses of neurotoxic astrocytes after SCI. In primary mouse astrocytes, we stimulated astrocytes using A1IM. The expression of C3, a marker of A1-like neurotoxic astrocytes (Alawieh et al., 2021), was particularly high in A1IM-stimulated astrocytes. Nevertheless, pre-treatment with RUX led to a gradual reduction in the expression of C3 in A1IM-stimulated astrocytes, and this effect was observed in a concentration-dependent manner. These findings provide clear evidence of RUX’s effectiveness in inhibiting the activation of neurotoxic astrocytes (Figure 8A and B).
To further substantiate this finding, astrocytes pre-treated with 1-μM RUX were stimulated with A1IM and analyzed via immunofluorescence staining. The results demonstrated that RUX significantly reduced the fluorescence intensity of C3 in A1IM-stimulated astrocytes (Figure 8C and D). Given the essential role of the NFκB pathway in inflammatory responses, we examined whether RUX could inhibit the phosphorylation of P65, a key transcription factor in the NFκB signaling pathway (Ren et al., 2022). Western blotting revealed that RUX markedly suppressed the phosphorylation of P65 in A1IM-stimulated astrocytes (Figure 8E and F). qPCR revealed that RUX significantly reduced the elevated mRNA levels of IL-6 and TNF-α induced by A1IM. Meanwhile, the decrease in IL-1β did not reach statistical significance (Figure 8G).