视神经损伤
-
Figure 2|HE staining assessment of retinal injury after blunt ocular trauma.
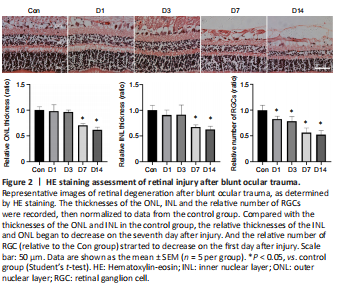
HE staining results showed that blunt ocular trauma induced retinal degeneration, rather than retinal detachment (Figure 2). The relative thicknesses of the ONL, INL, and GCL were calculated; the number of cells in the ONL, INL and GCL were compared between the injury and control groups (all P < 0.01). The relative number of RGCs sharply decreased by nearly 50% on the 14th day after injury, compared with the control group. The numbers of cells in the INL and ONL layers began to decrease on the seventh day after injury (P < 0.01). These findings suggest that blunt ocular trauma can lead to retinal injury and have an impact on ocular health.
Figure 3|Damage to the ONL and INL worsens on the seventh day after blunt ocular trauma.
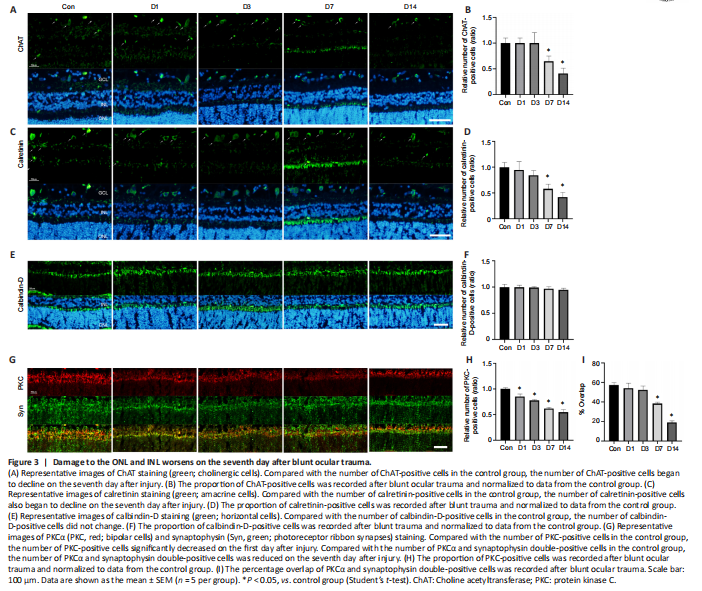
Various types of retinal cells responded differently to blunt ocular trauma in this study; the thicknesses of the ONL and INL decreased after injury. Rod and cone photoreceptor cells, which are the main components of the ONL, exhibited injury until the seventh day after blunt ocular trauma; the relative thickness of the ONL decreased over time (Figure 2). Next, immunofluorescence staining was performed to investigate the effects of blunt ocular trauma on horizontal, bipolar, Müller, and amacrine cells in the INL. Staining for ChAT (a marker of cholinergic cells) revealed that the number of cholinergic cells (a subset of amacrine cells) began to decline at 7 days after injury (P < 0.05; Figure 3A and B). The number of calretinin-positive cells (i.e., amacrine cells (Keilhoff et al., 2021)) also decreased at 7 days after injury (P < 0.01; Figure 3C and D). The number of calbindin-D-positive cells (i.e., horizontal cells) was unchanged after blunt ocular trauma (P > 0.05; Figure 3E and F); the relative number of calbindin-D-positive cells also did not change. Nevertheless, PKCα-positive cells (i.e., bipolar cells) appeared to be vulnerable to blunt ocular trauma, such that the number of bipolar cells significantly decreased on the first day after injury (P < 0.05; Figure 3G and H). Intersections of bipolar cells and synaptophysin-positive photoreceptor ribbon synapses are involved in visual transduction. We found that the connections between bipolar cells and photoreceptor cells were reduced on the seventh day after injury (P < 0.01; Figure 3G and I). Overall, these results suggest that bipolar cells were affected by blunt ocular trauma in a manner similar to rods and cones; with the exception of horizontal cells, all cells in the INL exhibited injury beginning at 7 days after blunt ocular trauma.
Figure 4|Blunt ocular trauma induces retinal cell necroptosis.
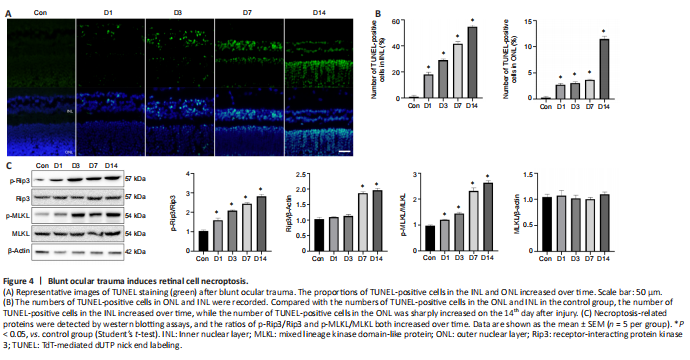
TUNEL assays showed that retinal cell death occurred on the first day after injury in the INL and ONL (both P < 0.01; Figure 4A and B), with dramatic increases in the number of TUNEL-positive cells. The number of TUNEL-positive cells in the INL increased over time, while the number of TUNEL-positive cells in the ONL was sharply increased at 14 days after injury (Figure 4A and B). The progression of retinal injury progressed from the GCL to the ONL (Figure 4A), suggesting that some detrimental factors such as ischemia, oxidative stress and inflammation, might be involved in the process of retinal injury and exacerbation of retinopathy. To investigate the role of necroptosis in blunt ocular trauma, we examined necroptosis-related proteins, such as p-Rip3, Rip3, p-MLKL, and MLKL. We found that the ratios of p-Rip3/Rip3 and p-MLKL/MLKL both increased over time (both P < 0.01; Figure 4C), suggesting that necroptosis plays an important role in the exacerbation of retinal injury.
Figure 5|Müller cell regeneration and gliosis occur after blunt ocular trauma.
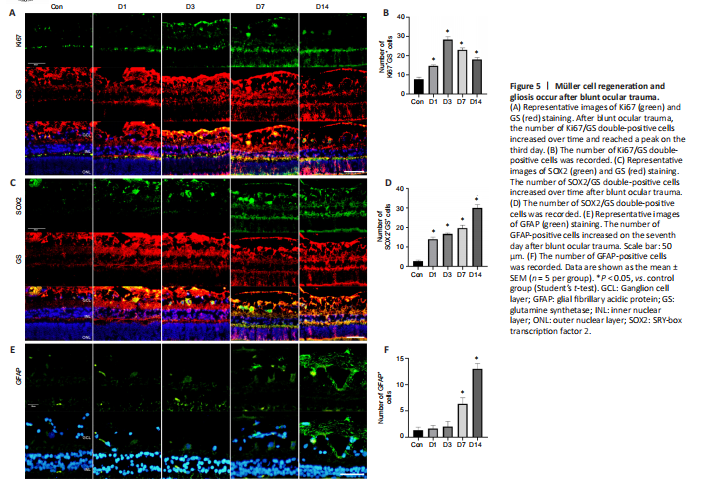
Double immunofluorescence staining of Ki67 and GS showed that the number of Ki67-positive Müller cells increased beginning on the first day after trauma and reached a peak on the third day after blunt ocular trauma (both P < 0.01; Figure 5A and B). SOX2 and GS staining showed that the number of SOX2-positive Müller cells increased beginning on the first day after blunt ocular trauma (both P < 0.01; Figure 5C and D); the relative number of Müller cells increased in a similar manner. Additionally, the number of GFAP-positive cells increased on the seventh day after injury (P < 0.01), suggesting enhanced Müller cell gliosis (Figure 5E and F). Thus, Müller cell gliosis was activated after Müller cell regeneration.
Figure 6|Inflammation and microglial polarization occur after blunt ocular trauma.
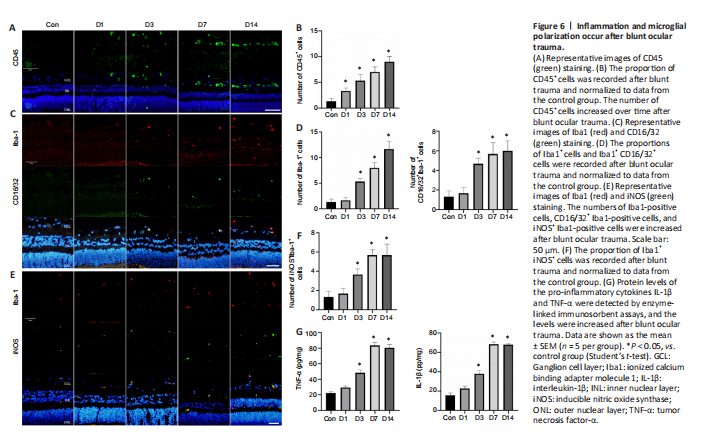
CD45 staining was performed to assess the inflammatory response; the relative number of CD45-positive cells in the injury group increased over time compared with that in the control group (both P < 0.05; Figure 6A and B). To investigate the role and characteristics of spatiotemporal changes in microglia after blunt ocular trauma, we performed immunofluorescence staining of microglia in the retina. We found that microglia were activated and recruited into the injured retina (Figure 6C–F), such that the relative number of Iba1-positive cells in the retina was elevated (P < 0.01). CD16/32 and iNOS are cellular markers of M1 microglia, which are presumed to secrete pro-inflammatory cytokines and promote neuroinflammation (Li et al., 2021a). The number of M1 microglia increased on the seventh day after injury (both P < 0.01; Figure 6C–F); this was accompanied by increased levels of the pro-inflammatory cytokines IL-1β and TNF-α after injury (both P < 0.01; Figure 6G). Moreover, the activation and polarization of microglia followed the pattern of progression from the GCL to the INL and ONL (Figure 6C–F), suggesting that the progression of retinal cell death is associated with microglial activation.