神经损伤与修复
-
Figure 5|Disruption of the plasma membrane integrity and Ca2+ dyshomeostasis evoked by αS oligomers and fibrils.
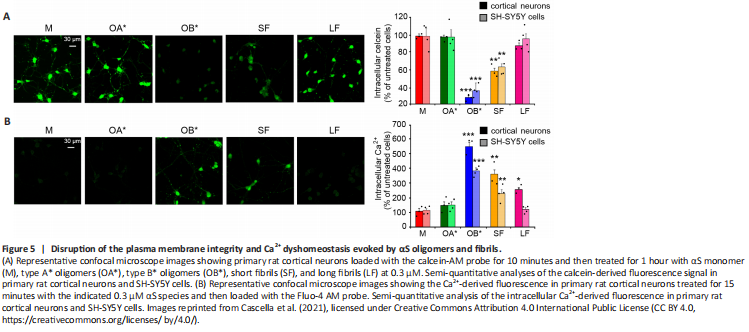
The maintenance of the plasma membrane integrity is of fundamental importance for every cell type, for normal functionality and survival. Its loss has been widely described as a cardinal pathogenic mechanism of PD and proteinopathies in general (Cenini et al., 2010; Evangelisti et al., 2016). αS aggregates, and in particular oligomers, were reported to exert their cytotoxic effects upon an abnormal interaction with lipid membranes (Iyer et al., 2019). Specifically, they can induce changes in the permeability and fluidity of the lipid bilayer, and influence the normal distribution of exposed proteins (Shrivastava et al., 2017). Their binding is generally mediated by their positively charged core, which establishes electrostatic interactions with negatively charged lipids present in the plasma membrane (St?ckl et al., 2012). This step is then followed by the permeabilization of the lipid bilayer, occurring through different mechanisms; the first one is the pore formation, or “poration” (Zakharov et al., 2007; Tosatto et al., 2012). This model assumes that annular αS oligomers with a central pore embed into lipid bilayers forming transbilayer proteins that allow the passage of small molecules and ions (St?ckl et al., 2013). Specifically, such pores were reported to possess an ion channel-like activity, resulting in an abnormal calcium influx (Figure 4; Danzer et al., 2007; Kim et al., 2009). Another possible mechanism is that in which αS oligomers cause the thinning of the lipid bilayer upon their binding, thus inducing an increase of membrane permeability and causing its leakage. According to this model, αS oligomers establish electrostatic interactions between their positively charged core and the negatively charged lipid headgroups; then, they are incorporated between the tightly packed lipids, thus evoking the thinning of the hydrophobic core of the bilayer and increasing its permeability (St?ckl et al., 2013). Finally, αS oligomers were also reported to bind to existing packing defects present in the bilayer, and extract phospholipids (Chaudhary et al., 2016); specifically, the increased exposure of lipid acyl chains at the edges of defects was reported to facilitate the interaction between oligomers and membranes, resulting in the growth of fractal domains in which there are no lipids (Chaudhary et al., 2016). Consistently, Reynolds and coworkers reported that the addition of monomeric wild-type and mutated A53T and E57K αS on the surface of supported lipid bilayers induced the protein adsorption to the membrane, whose subsequent aggregation caused both membrane thinning and lipid extraction in the near proximity of the growing aggregates (Reynolds et al., 2011). The previously described OB* and, to a minor extent, short fibrils (SF), were recently reported to alter the permeability of the membrane of synthetic synaptic-like small unilamellar vesicles and cultured neuronal cells, by inducing the release of the calcein-AM fluorescent probe (Cascella et al., 2021). A significant leakage of intracellular calcein, corresponding to the low green fluorescent signal in Figure 5A, was shown in rat primary cortical neurons following the addition to the culture medium of OB* and SF (Cascella et al., 2021), suggesting a permanent dysfunction of neuronal bilayers following the aggregates interaction with the lipid bilayer. Specifically, the propensity of OB* to permeabilize plasma membranes has been associated with their ability to anchor to the lipids through the N-terminus α-helical structure and to later insert their rigid β-sheet structure into the hydrophobic core of the membrane (Fusco et al., 2017). By contrast, all other αS species, such as αS monomer, OA* and long fibrils (LF), caused negligible calcein release (Figure 5A), indicating a low propensity to permeabilize neurons (Cascella et al., 2021).
Calcium (Ca2+) is an essential metal ion and the most ubiquitous intracellular second messenger, involved in the modulation of almost all neuronal processes, such as gene expression, synaptogenesis, synaptic transmission, energy production, membrane excitability, neuronal plasticity, learning and memory (Berridge, 1998; Kawamoto et al., 2012). Neuronal Ca2+ signaling depends both on highly transient and reversible elevation of cytosolic concentrations via an influx from the extracellular space through specific plasma membrane channels, as well as from its release from intracellular stores (Brini et al., 2014). αS aggregates, and particularly oligomers, were reported to enhance the plasma membrane permeability to Ca2+ ions through the perforation of the lipid bilayer by forming pore-like complexes (Danzer et al., 2007). Several observations have been performed after the addition of different αS species to the external medium of cultured cells. Angelova and coworkers revealed that both monomeric and oligomeric αS can induce a massive influx of Ca2+ ions in neurons and astrocytes, but only oligomers evoke neuronal cell death (Angelova et al., 2016). Consistently, we recently observed that a 15 min treatment with OB* and SF and, to a lesser extent, LF, evoked an extensive influx of Ca2+ both in cortical neurons and SH-SY5Y cells, whereas OA* and monomer caused a negligible elevation of intracellular calcium (Figure 5B; Cascella et al., 2021). Simultaneously, the interaction with a range of membrane channels and receptors, with the consequent modulation of their activity, has also been revealed. A prolonged treatment of rat hippocampal slices with αS oligomers, but not with the monomeric protein or fibrillar conformers, was demonstrated to cause a massive activation of N-methyl-D-aspartate receptors (NMDARs), which triggered in turn an increased activation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), so promoting the insertion into the postsynaptic membrane of higher conductance Ca2+-permeable versus GluR2-containing AMPARs, involved in basal synaptic transmission, ultimately leading to impaired long-term potentiation (Diógenes et al., 2012). More recently, Ferreira and coworkers revealed that the αS oligomers-induced impairment of long-term potentiation was due to the interaction with the cellular prion protein (PrPC), with the consequent phosphorylation of the Fyn kinase via metabotropic glutamate receptors 5, finally culminating in the phosphorylation and activation of NMDARs, through which a massive increase of intracellular Ca2+ levels occurs (Figure 4), ultimately triggering synaptic dysfunction in hippocampal neurons and synaptic and cognitive deficits in mice (Ferreira et al., 2017). More recently, Trudler et al. (2021) demonstrated that both oligomeric and fibrillar αS species evoke a rise in astrocytic intracellular Ca2+, which is responsible for a massive release of glutamate, that activates aberrantly extra-synaptic NMDARs on neighboring neurons, directly contributing to synapse loss. In the same work, αS oligomers were also reported to directly activate extra-synaptic NMDARs on neurons, thus exacerbating their damage (Trudler et al., 2021). Aggregated - but not monomeric - αS has also been shown to lead to Ca2+ dyshomeostasis upon intracellular accumulation; once in the cytoplasm, αS aggregates bind to the sarco/endoplasmic reticulum Ca2+-ATPase, thus stimulating its activity and triggering an initial reduction of cytosolic Ca2+ concentration and an overload in the ER (Figure 4). This phase is thought to be characterized by a significant imbalance of Ca2+-dependent processes, that is responsible for cellular dysfunction, and it is followed by a later increase in cytosolic Ca2+ levels, that precedes cell death (Betzer et al., 2018).