神经退行性病
-
Figure 1|CHCHD2 T61I mutation impairs mitochondrial function in a cell model of Parkinson’s disease.
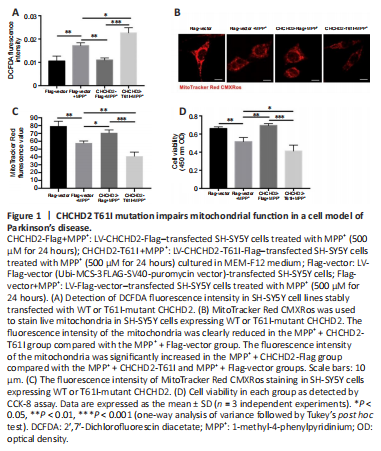
To investigate the role of WT CHCHD2 and T61I-mutant CHCHD2 in mitochondrial function, we detected cellular ROS levels by DCFDA assay. Cellular ROS levels in the MPP+ + Flag-vector group were significantly higher than those in the Flag-vector group (P < 0.01; Figure 1A). Furthermore, cellular ROS levels in the MPP+ + CHCHD2-Flag group were significantly lower than those in the MPP+ + Flag-vector (P < 0.01) and MPP+ + CHCHD2-T61I groups (P < 0.05; Figure 1A).
To investigate the effect of WT CHCHD2 and T61I-mutant CHCHD2 on mitochondrial activity, SH-SYSY cells were subjected to MitoTracker Red CMXRos staining. Mitochondrial fluorescence was clearly reduced in the MPP+ + CHCHD2-T61I group compared with the MPP+ + Flag-vector group (P < 0.01; Figure 1B and C). In addition, mitochondrial fluorescence intensity was significantly increased in the MPP+ + CHCHD2-Flag group compared with the MPP+ + CHCHD2-T61I (P < 0.001) and MPP+ + Flag-vector groups (P < 0.05; Figure 1B and C).
Next, cell viability was assessed by CCK-8 assay. Following MPP+ treatment, the viability of cells in the Flag-vector (P < 0.01) and CHCHD2-T61I SH-SY5Y groups (P < 0.001) was markedly lower than that in the CHCHD2-Flag group (Figure 1D). These results illustrate that CHCHD2 reduces cellular ROS levels and protects cells from oxidative stress injury and mitochondrial damage induced by T61I-mutant CHCHD2 and MPP+. This finding suggests that WT CHCHD2 plays a protective role and promotes cell survival in an MPP+-induced PD model.
Figure 3|CHCHD2 T61I mutation promotes mitochondrial permeability transition pore (mPTP) opening in SH-SY5Y cells.
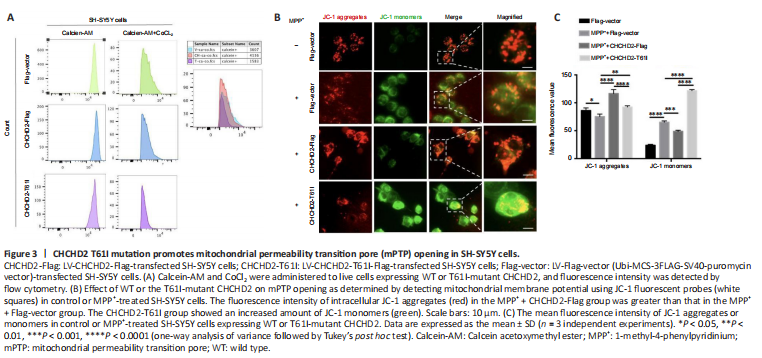
Figure 5|CHCHD2 T61I mutation aggravates movement deficits and nigral DA neuron function in a mouse model of PD.

Maintaining mPTP component integrity is crucial for ATP synthesis by mitochondrial F1F0-ATPase (Gauba et al., 2017). We assessed mPTP permeability by calcein-AM/cobalt staining and found that the degree of fluorescence quenching was greater in the CHCHD2-T61I group compared with the Flag-vector and CHCHD2-Flag groups (Figure 3A). The fluorescence in the cells incubated with ionomycin as a control was completely quenched (Additional Figure 4). These results imply that T61I-mutant CHCHD2 promotes pore opening and enhances mitochondrial permeability, while WT CHCHD2 protects mitochondria from developing excessive permeability.
We also assayed MMP using JC-1 fluorescent probes to detect the effect of WT CHCHD2 or the T61I mutation on mitochondria permeability. Under normal conditions, JC-1 stained as monomers indicative of a dissipated membrane potential (green) in the CHCHD2-T61I group, with almost no JC-1 monomers remaining in the Flag-vector and CHCHD2-Flag groups, suggesting that T61I CHCHD2 mutation could reduce the MMP and exacerbate the permeability of mitochondria (Additional Figure 5A and B). In MPP+ models, the number of JC-1 aggregates (red) in the cells of the MPP+ + CHCHD2-Flag group was increased than that of the MPP+ + Flag-vector group (P < 0.0001; Figure 3B and C). The CHCHD2-T61I group showed increased JC-1 monomers. This suggests that the MMP was decreased upon MPP+ treatment, and WT CHCHD2 but not the CHCHD2-T61I mutant could rescue the decreased MMP, where healthy mitochondria form JC-1 aggregates in matrix. The T61I CHCHD2 mutation may aggravate the opening of mPTP, and WT CHCHD2 may play a protective role against MPP+-induced mPTP opening.
To determine whether the CHCHD2 T61I mutation plays a role in PD pathogenesis, we overexpressed WT CHCHD2 or T61I-mutant CHCHD2 (via transfection with AAV-CHCHD2-Flag or AAV-CHCHD2-T61I-Flag, respectively) in a MPTP-induced mouse model of PD (Figure 5A). The animals in the AAV-CHCHD2-Flag group performed significantly better in the grasping test (P < 0.01; Figure 5B) and the pole-climbing test (P < 0.05; Figure 5C) than the animals in the AVV-Flag-vector group, while the animals in the AAV-CHCHD2-T61I group performed worse in both the pole-climbing test (P < 0.05; Figure 5C) and the rotarod test (P < 0.0001; Figure 5D). We observed a significant loss of tyrosine hydroxylase (TH)-positive neurons in the SN in the AAV-CHCHD2-T61I + MPTP group compared with the AAV-Flag-vector + MPTP and AAV-CHCHD2-Flag + MPTP groups, as assessed by immunohistochemistry assay (P < 0.01; Figure 5E and F), immunofluorescence staining (P < 0.01; Figure 5G and H), and western blotting (P < 0.05; Figure 5I and J). There were no significant changes in the grasping test and pole-climbing test results among the groups without MPTP treatment (Additional Figure 6A and B). Mice in the AAV-CHCHD2-T61I group performed distinctly worse than the AAV-Flag-vector group in the rotarod test (P < 0.0001; Additional Figure 6C). Immunohistochemical staining and western blot analysis showed a significant decrease in TH-positive neurons in the AAV-CHCHD2-T61I group compared with the AAV-Flag-vector (P < 0.05) and AAV-CHCHD2-Flag groups (P < 0.01; Additional Figure 6D–G). These findings indicate that the T61I CHCHD2 mutation can aggravate DA neuron degeneration and MPTP-induced motor functional deficit, and that WT CHCHD2 may protect dopaminergic neurons.
Figure 7|Interaction between CHCHD2 and OSCP in SH-SY5Y cells.
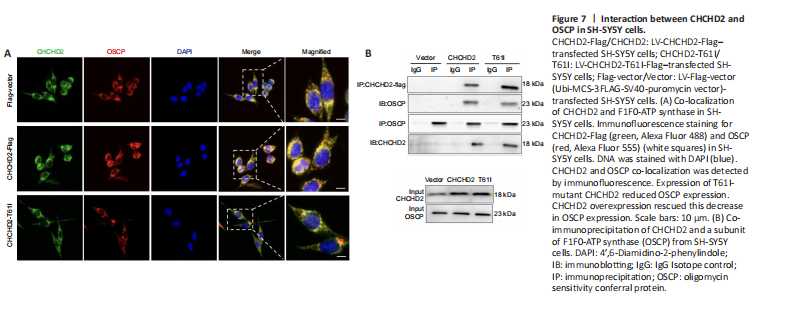
To determine how CHCHD2 regulates F1F0-ATPase assembly, we conducted LC-MS/MS protein profiling analysis and found that CHCHD2 specifically interacts with peptides belonging to the F1F0-ATPase subunit OSCP (Figure 6A–C). Immunofluorescence analysis demonstrated co-localization of CHCHD2 and OSCP (Figure 7A), and co-immunoprecipitation analysis showed that CHCHD2 interacts directly with OSCP (Figure 7B). These results suggest that CHCHD2 might target OSCP to modulate F1F0-ATPase function and assembly. Western blotting showed that CHCHD2 overexpression rescued the MPP+- or MPTP-induced decrease in OSCP expression in the in vitro or in vivo PD model, respectively (P < 0.05; Figure 8A–D). However, T61I-mutant CHCHD2 overexpression failed to increase OSCP expression in either model (P < 0.05; Figure 8A–D). There was no noticeable variability in OSCP expression among the control groups (P > 0.05; Additional Figure 7A–D). These findings suggest that the inability of T61I-mutant CHCHD2 to regulate OSCP expression levels may be a pathogenic mechanism involved in PD.