神经损伤与修复
-
Figure 2|3D structure of binding sites predicted by Phyre2 web tool.
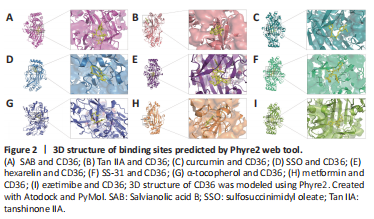
结果:SAB has been previously reported to target CD36, and surface plasmon resonance analysis showed that SAB directly bound to CD36. For this review, we performed molecular docking prediction for SAB and CD36 using Autodock Vina software (http://vina.scripps.edu; Eberhardt et al., 2021), and the results showed that SAB does have a binding spatial structure with CD36, which further explained the basis of its function (Figure 2A). One study found that SAB not only reduced oxLDL-induced CD36 expression in in vitro experiments but also inhibited CD36 in in vivo experiments (Bao et al., 2012a). Further experimental studies were conducted to determine whether SAB could inhibit CD36 in lipid metabolism-related neurological disorders and thus provide therapeutic benefits. Hyperlipidemia can exacerbate post-stroke injury with severe brain swelling, and researchers further investigated the role of CD36 inhibition in reducing infarct size and swelling in patients with hyperlipidemic stroke (Kim et al., 2020). Unfortunately, the mechanisms of action were not clarified in this study. However, it has been shown that SAB can improve metabolic function by inhibiting CD36 expression, thereby reducing visceral fat accumulation and improving insulin resistance in a wild-type mouse model of obesity (Yang et al., 2018). Thus, inhibition of CD36 by SAB may normalize metabolic function in patients with hyperlipidemia and reduce brain swelling after stroke. Furthermore, the loss of vascular tonus and BBB integrity underpins the pathology of stroke and AD, and stroke pathology also involves multiple factors that contribute to the death response, including inflammation and oxidative stress. CD36 has been used as a therapeutic target for endothelial dysfunction in stroke, where CD36-mediated pathways are activated by different ligands that will lead to inflammatory responses and endothelial dysfunction (Richards et al., 1994; Cho, 2012). SAB, as a CD36 antagonist, has been shown to inhibit the production of mitochondrial reactive oxygen species in endothelial cells and reduce oxLDL to achieve a protective effect in endothelial cell dysfunction (Ko et al., 2020). Therefore, the use of SAB to improve endothelial dysfunction and the inflammatory response is beneficial for alleviating stroke progression, but more experiments are needed to elucidate the exact mechanisms behind the inhibition of CD36 by SAB in neurological diseases. Notably, in neurological diseases not associated with lipid metabolism such as SCI, targeted inhibition of CD36 expression using SAB has been shown to attenuate CD36-mediated scar formation after SCI to reduce fibrosis, providing theoretical data for the treatment of spinal scars (Gong et al., 2023). In conclusion, SAB as an effective CD36 antagonist has potential for the treatment of neurodegenerative diseases and related complications by inhibiting CD36 function.
In a study on the role of Tan IIA in regulating CD36, researchers found that oxLDL mediated NLRP3 inflammasome activation via CD36. The use of Tan IIA reduced the expression of CD36 in macrophages and limited mitochondrial and lysosomal damage, eventually inhibiting the activation of NLRP3 inflammasomes and mitigating the inflammatory response (Zlowodzki et al., 2007; Wen et al., 2020). Therefore, inhibition of CD36 expression using Tan IIA to reduce NLRP3 inflammasome activation, systemic inflammation, and neuroinflammation is important to improve the symptoms of AD and stroke. Further analysis of the mechanisms by which Tan IIA inhibits CD36 expression was performed by Tang et al. (2011), who found downregulation of CD36 mRNA by Tan IIA (Figure 1B). We performed molecular docking prediction for Tan IIA with CD36 using Autodock Vina software, and the findings showed that Tan IIA has a spatial structure that links to CD36, which further explained the basis of its function (Figure 2B).
In a study on the regulation of CD36 expression by curcumin, Min et al. (2013) found that curcumin significantly inhibited CD36 expression in macrophages and that the expression and activity of peroxisome proliferator-activated receptor-γ (PPAR-γ), which is involved in the expression of CD36, was also inhibited in cells treated with curcumin. We also performed a molecular docking prediction of curcumin with CD36 using Autodock Vina software, and the results showed that curcumin does have a binding spatial structure with CD36, which further explains the basis of curcumin’s action (Figure 2C). It has been previously shown that CD36-mediated activation of NLRP3 inflammasomes leads to the accumulation of pro-inflammatory factors and that inhibition of CD36 expression attenuates NF-κB activation and prevents subsequent activation of the NLRP3 inflammasome, thereby reducing the inflammatory response (Jiang et al., 2021). Notably, in a model of ischemic stroke, curcumin was shown to improve functional outcomes by inhibiting NF-κB pathway activation and NLRP3 inflammasomes, ameliorating post-stroke white matter and brain tissue damage, and attenuating microglia apoptosis (Ran et al., 2021). Unfortunately, the mechanisms of CD36 involvement were not explored in this study, and more experiments are required to further elucidate the specific mechanisms by which curcumin binding to CD36 affects the NF-κB/NLRP3 pathway.
Sulfosuccinimidyl oleate (SSO) is a long-chain fatty acid with the molecular formula C22H36NNaO7S that has the ability to inhibit the oxidation of mitochondrial long-chain fatty acids, is an inhibitor of the mitochondrial respiratory chain, and is an antagonist of CD36 (Drahota et al., 2010). We also performed molecular docking prediction for SSO and CD36 using Autodock Vina software, and the findings showed that SSO has a binding spatial structure with CD36 (Figure 2D). Studies found that the target of SSO is located at lysine 164 on CD36 (Harmon and Abumrad, 1993; Kuda et al., 2013). In stroke, the post-ischemic inflammatory response is highly detrimental to recovery. Microglia and astrocytes are involved in this response, and microglia are rapidly activated and release pro-inflammatory mediators and cytotoxic substances after cerebral ischemia. Data have shown that CD36 is expressed on microglia and is involved in ischemic brain injury (Cho et al., 2005; Vidale et al., 2017; Qin et al., 2019).
CD36 is involved in mediating multiple signaling pathways that play an essential part in the pro-inflammatory response, including the NF-κB signaling pathway. One study found that NF-κB activity was diminished in CD36-deficient mice, suppressing the inflammatory response induced by cerebral ischemia and thereby reducing brain injury (Kunz et al., 2008). In another study, inhibition of CD36 expression using SSO blocked the activation of NF-κB and was able to prevent subsequent activation of inflammatory vesicles (Jiang et al., 2021). Thus, SSO may exert neuroprotective effects by binding to CD36. Interestingly, SSO was found to inhibit inflammation independently of CD36 in ischemic stroke. The experimental results showed that SSO could directly reduce lipopolysaccharide/interferon-γ-induced pro-inflammatory mediators produced by microglia, such as interleukin-6 and tumor necrosis factor-α, and could prevent inflammation-induced neuronal death and attenuate ischemic brain injury with neuroprotective effects in a mouse model of ischemic stroke (Dhungana et al., 2017). In conclusion, SSO has an anti-inflammatory profile that could make it a candidate molecule for further drug development. However, more studies are required to determine the exact mechanisms behind the neuroprotective effects of SSO inhibition of CD36 expression after ischemic stroke.
In addition to the development of new synthetic compounds as high-affinity scavenger receptor inhibitors, it has been shown that synthetic ligands of CD36 also show competitive binding properties with CD36. Hexarelin is a synthetic peptide with growth hormone-releasing activity that binds to the CD36 receptor. Demers et al. (2004) found that the binding domain of hexarelin on CD36 overlaps with the binding domain of oxLDL on CD36, and hexarelin may thus interfere with CD36-mediated uptake of oxLDL. We also performed molecular docking predictions for hexarelin with CD36 using Autodock Vina software, and the results showed that hexarelin has a binding spatial structure with CD36 (Figure 2E). In addition, hexarelin has shown neuroprotective and anti-apoptotic effects; hexarelin could protect Neuro-2a cells from H2O2-induced injury via a molecular pathway involving mitogen-activated protein kinase (MAPK). The experimental use of hexarelin attenuated H2O2 stimulation-induced MAPK upregulation (Meanti et al., 2021). Data showed that CD36 plays a key role in activating the MAPK pathway and that inhibition of MAPK signaling reduces apoptosis and contributes to neuroprotection (Kwon et al., 2011; Sini et al., 2017). Oxidative stress is involved in the pathogenesis of neurological diseases such as stroke, AD, and PD, and oxLDL levels influence stroke prognosis. Therefore, inhibition of CD36-mediated pathways using hexarelin has potential therapeutic relevance in the treatment of CNS diseases (Cho, 2012; Wang et al., 2017).
CD36 has recently been identified as a multimodal target for reducing oxidative stress and inflammation in ischemic stroke. In a study on the targeted inhibition of CD36 by SS-31, Zhang et al. (2017b) found that injection of SS-31 downregulated CD36 expression, inhibited oxidative stress, and improved systemic inflammation in mice. We performed molecular docking prediction for SS-31 and CD36 using Autodock Vina software, and the results showed a binding spatial structure between SS-31 and CD36 (Figure 2F). Cho et al. (2007) showed that treatment with SS-31 in a C57BL/6 mouse model of cerebral artery occlusion inhibited CD36 expression, reduced inflammatory cells and infarct size, and had neuroprotective effects. Notably, use of the SS-31 to target CD36 in the treatment of ischemic stroke was effective in a transient stroke model but virtually ineffective in a permanent ischemic stroke model (Kim et al., 2015). Therefore, targeted inhibition of CD36 by SS-31, a novel antioxidant, may be a useful therapeutic strategy to reduce acute stroke-induced injury and has potential therapeutic relevance in neurodegenerative diseases (Figure 1B).
Vitamin E is one of the most prominent antioxidants and is an essential nutrient for neurodevelopment and cognitive function; vitamin E deficiency leads to neurodegenerative diseases such as ataxia and cognitive decline (Traber, 2021). α-Tocopherol is the most active form of vitamin E and was found to have a regulatory function on CD36. Ricciarelli et al. (2000) found in in vitro experiments that α-tocopherol could downregulate CD36 mRNA expression and protein translation by reducing CD36 promoter activity, thereby inhibiting oxLDL uptake. We performed molecular docking prediction for α-tocopherol and CD36 using Autodock Vina software, and the results showed a binding spatial structure between α-tocopherol and CD36 (Figure 2G). In a recent study, Zingg et al. (2022) used vitamin E analogues and derivatives, and found that both α-tocopherol phosphate (αTP) and an αTP/β-cyclodextrin nanocarrier complex inhibited CD36 surface exposure in human acute monocytic leukemia cells, leading to decreased oxLDL uptake. Similarly, interferon-γ was reported to significantly inhibit cell surface CD36 expression in a dose-dependent manner and to stimulate macrophages, leading to the synthesis of 7,8-dihydroneopterin, a potent antioxidant that inhibits cellular oxidative damage (Nakagawa et al., 1998). The reason for this was investigated by Gieseg et al. (2010), who showed that the reduction in oxLDL uptake was due to a significant downregulation of CD36 levels. Studies found that, similar to α-tocopherol, 7,8-dihydroneopterin inhibited CD36 expression via a cellular signaling pathway that is also associated with PPAR-γ (Munteanu et al., 2006; Ghodsian et al., 2022). In addition, another antioxidant, N-acetylcysteine, has also shown an ability to inhibit CD36 expression (Ding et al., 2022). Notably, antioxidants that inhibit CD36 expression can be used to reduce oxLDL levels as well as attenuate oxidative stress to have neuroprotective effects and, thus, improve prognosis in stroke (Murad et al., 2014). However, data suggest that excessive antioxidant levels can exacerbate the neuroinflammatory response and oxidative stress in acute ischemic stroke, ultimately leading to increased brain damage (Khanna et al., 2015). Therefore, antioxidant dosages should be carefully considered in the treatment of people at high risk of stroke.
Some drugs, such as AP5055, AP5258, ezetimibe, metformin, and statins, have regulatory effects on CD36 expression (Geloen et al., 2012; Zeng et al., 2019; Yu et al., 2020). Rekhi et al. (2021) further showed that AP5055 could target CD36 and reduce NF-κB activation, thereby reducing interleukin-1β production and ultimately reducing the inflammatory response. Ezetimibe is a selective inhibitor of cholesterol uptake and is commonly used in clinical practice for lipid-lowering therapy. Qin et al. (2016) showed that ezetimibe also has reduced CD36 expression. Further, metformin also has inhibitory effects on CD36 (Moon et al., 2017). Metformin inhibits NF-κB translocation in macrophages, which lowers the synthesis of pro-inflammatory cytokines as well as the expression of the scavenger receptor CD36 in macrophages, both of which work together to lower inflammation (Hyun et al., 2013). We used Autodock Vina software to conduct molecular docking prediction for ezetimibe and metformin with CD36, and the results showed that ezetimibe and metformin both had a binding spatial structure with CD36 (Figure 2H and I). Studies have also shown that statins downregulated CD36 expression in in vivo experiments and reduced inflammatory factors such as interleukin-6 and tumor necrosis factor-α (Yin et al., 2017). Regarding the mechanisms of CD36 downregulation by simvastatin, experiments showed that inhibition of calpain-1 may mediate the downregulation of PPAR-γ by simvastatin, thereby inhibiting CD36 mRNA expression (Yang et al., 2016). Berberine is a novel lipid-lowering drug, and it has been reported that berberine can reduce the expression of CD36 mRNA (Sun et al., 2017). The main active metabolite of berberine, berberrubine, can also regulate CD36 expression to prevent lipid accumulation (Yang et al., 2022). Interestingly, during the synthesis of berberine analogues, researchers also accidentally discovered the generation of an N-(2-arylethyl) isoquinoline derivative and screened its activity. It was also found to have good activity in inhibiting CD36 and was considered as a promising new CD36 antagonist (Wang et al., 2011). A new strategy based on a family of “nanoblockers” was also examined by Chnari et al. (2006). They created nanoblockers that targeted the scavenger receptors SR-A and CD36 and showed that doing so efficiently prevented the uptake of oxLDL. Doens et al. (2017) reported the development of a colorimetric assay for screening molecules that interfere with CD36-Aβ interactions. Using this assay, seven compounds were experimentally identified that interfered with the binding of Aβ to CD36. By utilizing pharmacological inhibitors of CD36-Aβ interactions, new potential therapeutic agents for the treatment of AD may be obtained.
点击此处查看全文