神经退行性病
-
Figure 1|Expression and subcellular localization of Atg9 in adult fly brain glia.
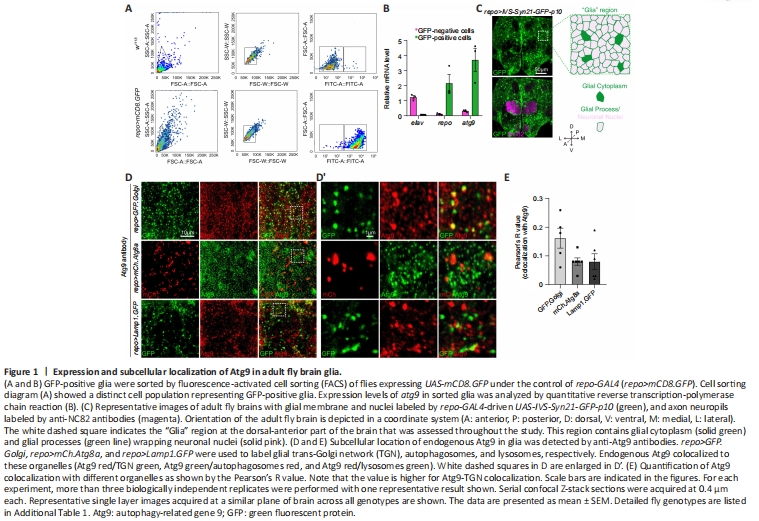
To determine if Atg9 plays a role in glial cells, we first labeled adult fly brain glia by crossing UAS-mCD8.GFP to a pan-glial driver repo-GAL4, which is expressed in all types of glial cells (repo>mCD8.GFP). These labeled glial cells were first sorted by fluorescence-activated cell sorting and then analyzed for atg9 expression via qRT-PCR (Figure 1A and B). Sorted GFP-positive cells had high repo (glial-specific marker) and low elav (neuron-specific marker) expression, indicative of purified glial cells. Interestingly, atg9 expression was much higher in GFP-positive cells than GFP-negative cells (Figure 1B), indicating that Atg9 might play a role in glia.
Next, the adult Drosophila brain was analyzed by immunostaining with different markers. The overall structure and location of different brain cell types were characterized with anti-NC82 antibodies (magenta, neuropil) and the glia-specific repo-GAL4 driven UAS-IVS-Syn21-GFP-p10 (green, glial membrane and nuclei) (Figure 1C). Image analysis showed that glial cells were widely distributed in the adult fly brain, with some glia terminal processes in close proximity to the axonal neuropil. A “Glia” region at the dorsal-anterior part of the brain (which contains glial cytoplasm and glial processes wrapping neuronal nuclei) was chosen for further analysis.
During Drosophila development, Atg9 was reported to co-localize with Atg8a-labeled autophagosomes, Rab11-labeled endosomes, and Lamp1-labeled endosomes-lysosomes in the fat body (Bader et al., 2015). We therefore checked whether Atg9 has a similar subcellular location in adult fly brain glia. Transgenic flies (UAS-GFP.Golgi, UAS-mCherry.Atg8a, and UAS-Lamp1.GFP) were crossed with repo-GAL4 flies to mark TGN, autophagosomes, and lysosomes in glia, respectively. Endogenous Atg9 was detected with anti-Atg9 antibodies, with specificity validated by atg9-RNAi (Additional Figure 1A and B). Consistent with previous studies demonstrating Atg9 localization in other tissues, Atg9 also localized to TGN, autophagosomes, and lysosomes in adult fly glia, with a higher portion of protein localizing to TGN (Figure 1D and E).
Figure 2|Trafficking dynamics of Atg9 in glial cells.
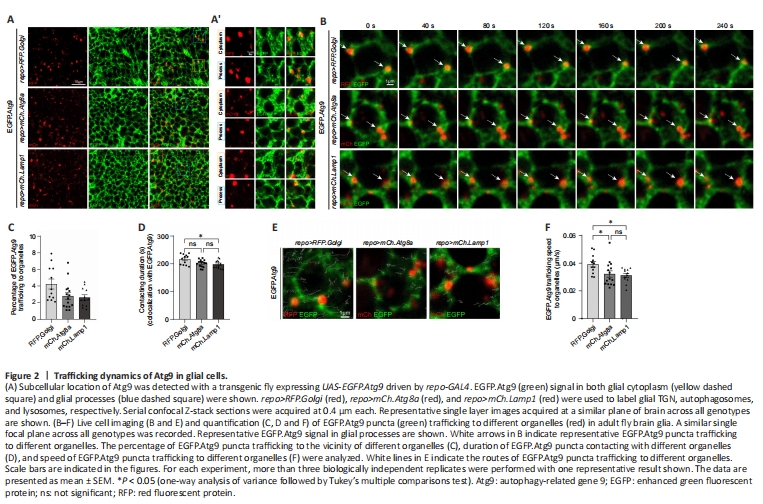
Alternatively, a transgenic fly line expressing EGFP.Atg9 (UAS-EGFP.Atg9) was created and crossed to repo-GAL4 for detecting Atg9 expression in glia. EGFP.Atg9 expression and its colocalization with TGN, autophagosomes, or lysosomes were analyzed using different markers labeling these organelles. Under overexpression conditions, EGFP.Atg9, in both the glial cytoplasm and glial processes, showed obvious colocalization with TGN, autophagosomes, and lysosomes (Figure 2A and 2A’). Together with the above results, glial Atg9 may function in autophagy, as in other tissues.
To further analyze the dynamics of Atg9 on different organelles, live cell imaging of adult fly glia expressing EGFP.Atg9 in the presence of marker-labeled TGN, autophagosomes, or lysosomes was conducted (Figure 2B and Additional Videos 1–3). The percentage of EGFP.Atg9 trafficked to the vicinity of these organelles, their contacting time, and speed were quantified. Notably, the percentage of EGFP.Atg9 puncta trafficking to the vicinity of Golgi puncta was higher than the number of Atg9 trafficking to autophagosomes or lysosomes. Furthermore, Atg9-Golgi contacting duration was also longer than Atg9-autophagosome or Atg9-lysosome contacting duration (Figure 2B–D). Meanwhile, based on the trafficking route of EGFP.Atg9 puncta (Figure 2E), the speed of these puncta trafficking to TGN, autophagosomes, or lysosomes was calculated separately. Interestingly, Atg9 is most mobile when trafficking to TGN (Figure 2E and F). These observations show that Atg9 is trafficked to TGN, autophagosomes, or lysosomes in glia, yet more Atg9 is trafficked to TGN with a faster speed and longer contacting duration.
Figure 3| Glial Atg9 regulates omegasome and autophagosome formation and substrate degradation.
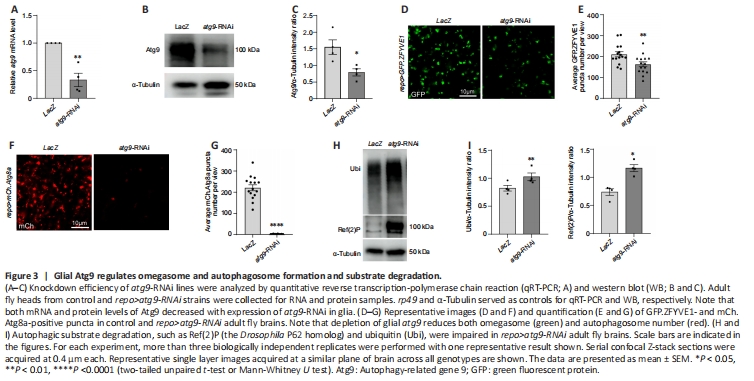
It has been shown that ATG9/Atg9 regulates the early steps of autophagy, and is the main membrane source for omegasome and autophagosome formation (Lang et al., 2000; Noda et al., 2000; Reggiori et al., 2004; Mari et al., 2010; Orsi et al., 2012; Yamamoto et al., 2012). Because we found that atg9 has high expression level in glial cells, we then determined whether Atg9 has similar functions in glia autophagy. Next, atg9 was inhibited by crossing atg9-RNAi (either atg9-RNAi BL#28055 or atg9-RNAi#2 V#10045) to repo-GAL4. Then GFP.ZFYVE1-labeled omegasomes (Liu et al., 2018) and mCh.Atg8a-labeled autophagosomes were quantified when atg9 expression was inhibited in glia. UAS-LacZ was used as a control. The knockdown efficiency of atg9-RNAi was validated by qRT-PCR, western blot (WB), and immunostaining (Figure 3A–C and Additional Figure 1A–C). Notably, lack of glial atg9 markedly reduced the number of omegasomes (Figure 3D and E) and autophagosomes (Figure 3F and G, Additional Figure 1D and E). These results show that glial Atg9 regulates autophagy by affecting the number of early autophagic structures such as omegasomes and autophagosomes.
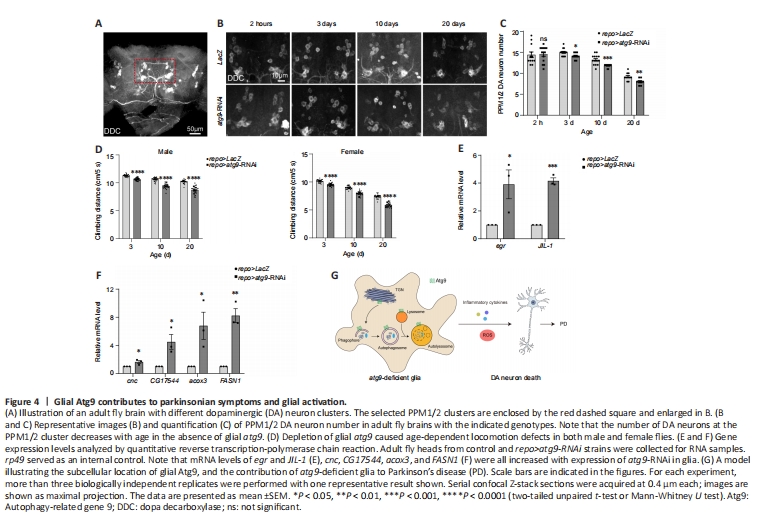
Figure 4| Glial Atg9 contributes to parkinsonian symptoms and glial activation.
Given that Atg9 regulates omegasome and autophagosome number, we next sought to investigate if the Atg9-mediated mechanism affects the efficiency of glial autophagy. Results from both WB and immunostaining indicated that the levels of autophagic substrates, Ref(2)P (the Drosophila P62 ortholog) and ubiquitin (Ubi), accumulated substantially when glial atg9 was depleted (Figure 3H and I, Additional Figure 1F and G), suggesting that Atg9 contributes to autophagic substrate degradation in glia.
As ATG9 dysregulation has been implicated in PD (Winslow et al., 2010; Yang et al., 2022), we then assessed whether the Atg9-mediated autophagic defects in glia play a role in PD using a Drosophila PD model (Song et al., 2017). DA neurodegeneration was detected by anti-DDC antibodies (Figure 4A). Notably, lack of glial atg9 caused progressive loss of the protocerebral posterior medial 1/2 (PPM1/2) DA neuron cluster, which has been shown to degenerate upon α-syn overexpression (Feany and Bender, 2000), in an age-dependent manner (Figure 4B and C). DA neuron number was not affected by the absence of glial atg9 immediately after eclosion, ruling out developmental defects (Figure 4B and C). Furthermore, fly climbing ability were quantified by an automated rapid iterative negative geotaxis assay (Cao et al., 2017; Song et al., 2017). Lack of glial atg9 caused age-dependent locomotion defects (Figure 4D). Thus, our results reveal that glial Atg9 regulates DA neurodegeneration and locomotor function, both typical hallmarks of PD (Song et al., 2017).
Numerous studies in patients with PD and animal models have shown that glial activation and neuroinflammation are new features of PD, which in turn exert neuroprotective or neurotoxic feedback to DA neurons (Ho, 2019; Badanjak et al., 2021; Wang et al., 2022; Yi et al., 2022). Activated glia release pro-inflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin (IL)-12, and IL-1β, establishing a chronic inflammatory milieu to exacerbate DA neurodegeneration (Ho, 2019; Wang et al., 2022; Yi et al., 2022; Zahedipour et al., 2022; Yu et al., 2023). We therefore checked whether pro-inflammatory cytokine expression was changed. qRT-PCR results showed that expression levels of egr and JIL-1, the Drosophila homologs of TNF-α and mitogen- and stress-activated protein kinase (MSK), were increased, suggesting increased pro-inflammatory cytokine expression (Figure 4E and Additional Figure 1H).
On the other hand, activated glia can induce reactive oxygen species (ROS) production, accelerating DA neurodegeneration (Ho, 2019; Wang et al., 2022; Yi et al., 2022). We also observed enhanced cnc expression, the nuclear factor erythroid 2-related factor 2 (Nrf2) homolog (Figure 4F and Additional Figure 1I). Moreover, expression levels of CG17544, acox3, and FASN1 were all increased, indicating increased ROS and lipid oxidation (Figure 4F and Additional Figure 1I). Taken together, these data suggest that loss of glial atg9 induces glial activation, which promotes DA neurodegeneration by releasing pro-inflammatory cytokines and ROS.
In the present study, we studied the glial function of the autophagy protein, Atg9, and its contribution to PD. We found that atg9 is highly expressed in glial cells from adult fly brain. Combining immunostaining and live cell imaging, we revealed that Atg9 is diversely localized in TGN, autophagosomes, and lysosomes in glia. In addition, Atg9 makes persistent contacts with these organelles. Consistent to Atg9 function in the fat body, Atg9 functions in glia by regulating the formation of omegasomes and autophagosomes, hence the degradation of autophagic substrates. Thus, glial Atg9 participates in the early steps of autophagy and controls the efficiency of autophagic degradation. As ATG9 dysregulation has been implicated in PD (Winslow et al., 2010; Yang et al., 2022), we then explored the effect of Atg9 in glia using a Drosophila PD model. Importantly, loss of glial atg9 induced parkinsonian symptoms including progressive loss of DA neurons and locomotion deficits. Moreover, loss of glial atg9 induced glial activation with greater release of inflammatory cytokines and ROS production, which in turn accelerates DA neurodegeneration. Taken together, our findings suggest that Atg9 modulates autophagic function in glia, and loss of glial atg9 contributes to PD (Figure 4G).