脑损伤
-
Figure 1 | CNH reduces gliosis, axon damage, and tauopathy after traumatic brain injury.
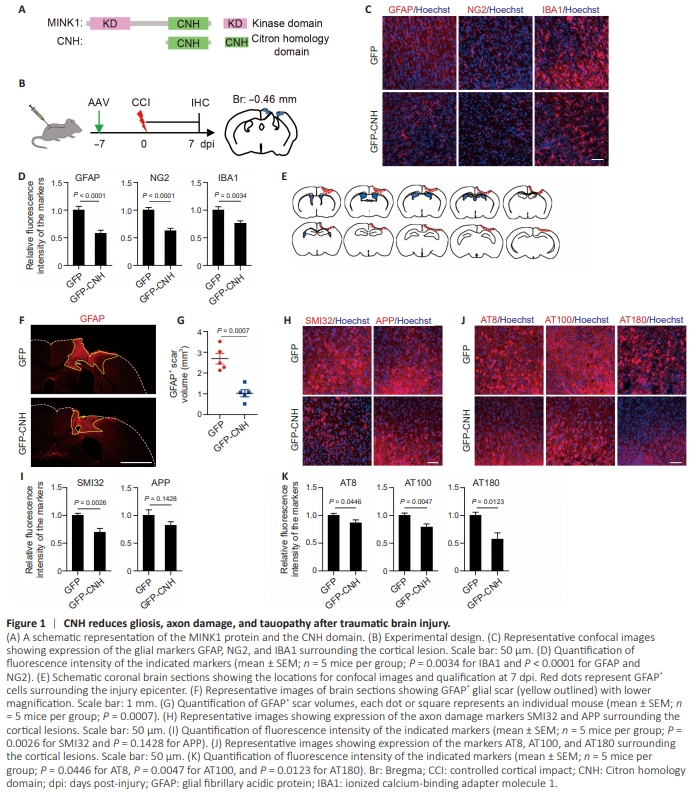
We first examined the cellular responses to CCI, a widely employed and highly reproducible mouse model of traumaticbrain injury (Onyszchuk et al., 2007; Chen et al., 2014; Kuwar et al., 2019). When the injured cortical area was analyzed at 7 days post-injury (dpi) and compared with the sham controls (Additional Figure 1A), CCI resulted in severe reactive gliosis indicated by dramatic increases of expression of GFAP (astrocytes), NG2 (NG2 glia), and IBA1 (microglia) (Additional Figure 1B). The injury also caused a remarkable upregulation of markers for axonal damage including SMI32 (a monoclonal antibody for detecting dephosphorylated neurofilaments) and amyloid precursor protein (APP) (Xie et al., 2010; Additional Figure 1B). We further examined hyperphosphorylated tau (p-tau) with the antibodies AT8 (for pS202/T205), AT100 (for pT212/S214), and AT180 (for pT231) as biomarkers of TBI (Uryu et al., 2007; Albayram et al., 2017; Rubenstein et al., 2017). In contrast to sham controls, CCI induced very extensive but dynamic p-tau expression, with the highest level at 4 dpi and gradual reduction to the basal level one month later (Additional Figure 1C and D). Together, these results confirm that TBI causes robust reactive gliosis, neurodegeneration, and tau pathology (Ng and Lee, 2019). The CNH domain ameliorates brain injury–induced pathology To determine whether TBI-induced pathology could be molecularly modulated, we focused on the STE20 family kinases MAP4Ks due to their broad brain expression (Chuang et al., 2016) and their critical roles in gliosis and neurodegeneration (Larhammar et al., 2017). Furthermore, it was recently shown that the CNH domain (henceforth referred to as CNH) of MINK1 (Figure 1A) could serve as a dominantnegative form blocking MAP4Ks to promote survival and function of human patient-derived neurons (Liu et al., 2024). This result raised the possibility of using CNH as a potential gene therapy for neural injury or degeneration. To test this possibility, we prepared AAVs to express GFP-CNH under the constitutively active CAG promoter. GFP alone was used as a control. AAVs were packaged with the PHP.eB capsid for efficient and brain-wide distributions (Mathiesen et al., 2020). Adult wild-type mice were intrathecally injected with AAVs and subjected to CCI 7 days post-virus (Figure 1B). When examined at 7 dpi and compared to the GFP control, the CNH group showed a significant reduction of reactive gliosis, measured by the relative expression of GFAP, NG2, and IBA1 surrounding the cortical injury (Figure 1C and D; P < 0.0001 for GFAP, P < 0.0001 for NG2, and P = 0.0034 for IBA1). Accordingly, the injury-induced glial scars, quantified by the volume of GFAP+ area, were greatly reduced in the CNH group (Figure 1E–G; P = 0.0007). CNH expression similarly resulted in a significant reduction of the axonal damage marker SMI32 and a trend of decreased expression of the injury-induced APP (Figure 1H and I; P = 0.0026 for SMI32 and P = 0.1428 for APP). The injury-induced hyperphosphorylated tau was also markedly dampened by the CNH expression compared with the control GFP (Figure 1J and K; P = 0.0446 for AT8, P = 0.0047 for AT100, and P = 0.0123 for AT180). Together, these results indicate that AAV-mediated expression of the CNH domain of MINK1 can broadly suppress brain injury-induced pathology including gliosis, neuronal damage, and tau pathology.
Figure 2 | CNH promotes tissue remodeling and behavior recovery after traumatic brain injury.
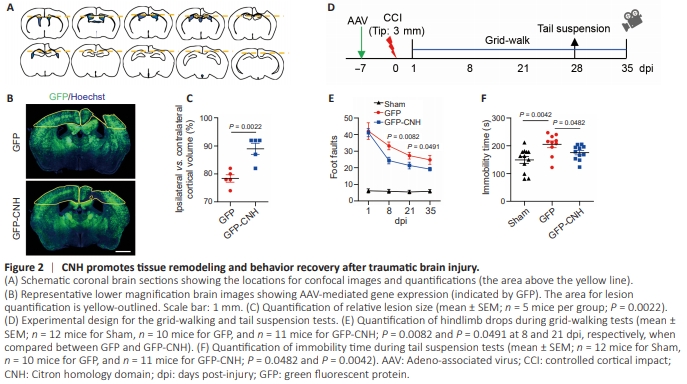
Supported by these above results, we next examined whether the CNH domain had any effect on tissue remodeling after TBI. Compared with the GFP control after CCI, the CNH group showed a much-reduced lesion size that was measured by the ratio of tissue area in the ipsilateral cortex to the contralateral cortex (Figure 2A–C; P = 0.0022). The functional impact of CNH on animals was then determined by behaviors after TBI. A cohort of adult wild-type mice were injected with AAVs, subjected to severe CCI, and examined by a battery of behavioral tests (Figure 2D). Since impaired locomotion is a sequela of TBI (Onyszchuk et al., 2007), animals were evaluated through a time course with the grid walking test, which quantifies foot faults made by the affected side (contralateral to the injury) when animals are allowed to walk on an elevated and even-spaced wire grid. This type of test is especially useful for models of unilateral TBI. Compared with sham animals at one dpi, both the CNH group and the GFP control exhibited near identical impairments indicating relatively uniform injuries among these mice (Figure 2E; P = 0.8662). However, the CNH group improved their motor function at a significantly faster pace than the GFP control in subsequent days (Figure 2E; P = 0.0082 at 8 dpi and P = 0.0491 at 21 dpi). Another TBI-induced impairment of brain function is depressive-like behavior (Carter et al., 2001; Chen et al., 2019), which was monitored by a tail-suspension test at 28 dpi (Figure 2D). Compared with the GFP control, the CNH group spent significantly less time in immobility indicating much-reduced depressive-like behavior (Figure 2F; P = 0.0482). Together, these results show that the CNH domain can improve functional recovery after TBI.
Figure 3 | CNH in neurons exerts neuroprotection.
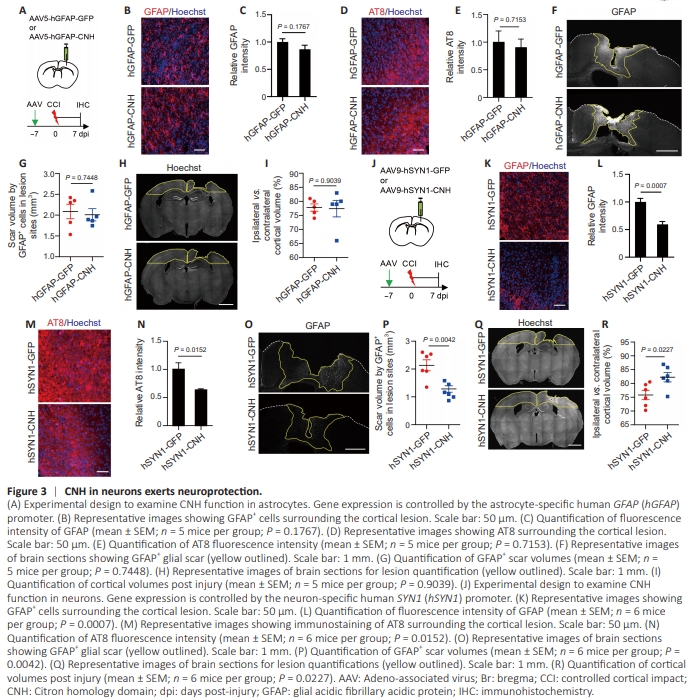
Since PHP.eB-serotyped AAVs can efficiently transduce both neurons and astrocytes (Chan et al., 2017), we next examined the cell type in which the CNH domain exerted its function. To target astrocytes, we used the human GFAP (hGFAP) promoter to drive gene expression and the AAV5 capsid to package AAVs. Consistent with the previous report (Wang et al., 2021), immunohistochemistry showed about 92% of the GFP+ cells were GFAP+ astrocytes in the mouse cortex that were transduced with the AAV5-hGFAP-GFP virus (Additional Figure 2A). When analyzed at 7 dpi and compared to the GFP control (Figure 3A), however, we failed to detect a significant effect of the astrocyte-expressed CNH domain on either reactive gliosis (indicated by the relative GFAP expression; Figure 3B and C; P = 0.1767) or tau pathology (indicated by the AT8 staining; Figure 3D and E; P = 0.7153). The astrocyte-expressed CNH domain was also unable to reduce glial scars (indicated by the GFAP+ cortical area; Figure 3F and G; P = 0.7448) or brain lesion size (indicated by the relative remaining cortical tissues; Figure 3H and I; P = 0.9039) after TBI. To target neurons, we used the human synapsin I promoter (hSYN1) to drive gene expression and the AAV9 capsid to package AAVs (Wang et al., 2021). Immunohistochemistry confirmed that about 94% of GFP+ cells were detected in NeuN+ neurons in the control AAV9-hSYN1-GFP virustransduced cortex (Additional Figure 2B). When examined at 7 dpi (Figure 3J), neuronal expression of the CNH domainled to a significant reduction of TBI-associated reactive gliosis (Figure 3K and L; P = 0.0007) and tau pathology (Figure 3M and N; P = 0.0152). It also resulted in greatly reduced glial scars and the lesioned cortical size (Figure 3O–R; P = 0.0042 for scar volume and P = 0.0227 for lesion size). Together, these results indicate that the CNH domain exerts its neuroprotective function predominantly in neurons but not in astrocytes after brain injury.
Figure 4 | Pharmacological inhibition of MAP4Ks is neuroprotective.

The recent results indicate that CNH works as a dominantnegative form to block MAP4Ks for improved survival and functions of human patient-derived neurons (Liu et al., 2024). To determine whether blocking MAP4Ks could exert a similar protective effect after TBI, we took a pharmacological approach. K02288 (Figure 4A; Sanvitale et al., 2013), also known as Hit3after chemical screens in human diseased neurons (Liu et al., 2024), is a potent inhibitor of MAP4Ks (Sanvitale et al., 2013; Liu et al., 2024). It was administered daily to mice for 7 days after CCI (Figure 4B). Compared with vehicle-treated mice, K02288 treatments markedly lowered reactive gliosis indicated by substantial reductions of the markers for astrocytes (GFAP+ ), NG2 glia (NG2+ ), and reactive microglia/macrophages (CD45+ ) after CCI (Figure 4C and D; P < 0.0001 for GFAP, P = 0.0192 for NG2, P = 0.0089 for CD45). Significant reductions were similarly observed for markers of neuronal damage (SMI32+ and APP+ ) and tau pathology (AT8 and AT100) in mice treated with K02288 after CCI ( Figure 4E and F; P = 0.0193 for SMI32, P = 0.0118 for APP, P = 0.0351 for AT8). Consequently, K02288- treated mice exhibited much reduced scar volume and lesion size (Figure 4G–J; P = 0.0092 and P = 0.0486, respectively). Together, these above results indicate that pharmacological inhibition of MAP4Ks is neuroprotective, consistent with the functions of AAV-CNH in vivo.