神经损伤与修复
-
Figure 1 | Only partial and transient neuritogenesis is induced by different agents in NSC-34 cells.
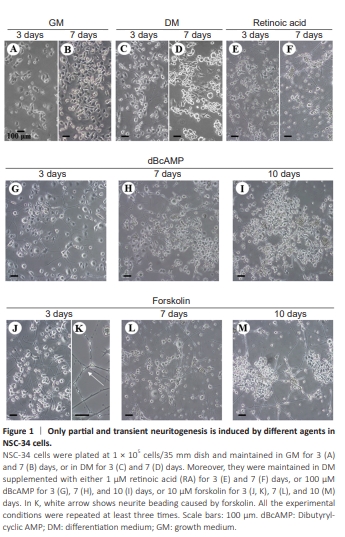
We showed that NSC-34 cells grown in GM were heterogeneous in shape and exhibited variable proportions of spherical or irregularly flat cells bearing short spikes (Figure 1A and B). When cultured in DM, only very few cells elongated cellular processes that reached up to 5–8 cell bodies in length, by approximately day 3 (Figure 1C). After 7 days, these protrusions however withdrew, and the cells started duplicating again in culture (Figure 1D). By confirming previous results (Maier et al., 2013), we showed that RA (1 μM) potentiated not only the length of these processes, but also the percentage of neurite-bearing cells, which reached up to nearly 50% of the entire population, on day 3 (Figure 1E). However, on day 7 we confirmed that RA became highly toxic, causing cytoplasmic vacuolation, massive cell death, and debris, together with beading, retraction and clear deterioration of the neuritic branches (Figure 1F).
To investigate the mechanisms related to the ara-C-induced differentiation of NSC-34 cells, we adopted a pharmacological approach and tested some DNA/RNA intercalating agents. Actinomycin D inhibiting DNA-primed RNA synthesis by blocking rRNA and heterogeneous nuclear RNA production, and cycloheximide blocking tRNA binding and release from ribosomes, both interfered with neuritogenesis when used at 10–1000 ng/mL and 1–5000 ng/mL respectively, thus suggesting that novel transcription and protein synthesis are indeed required for maintaining healthy neurites. In particular, at the highest tested concentration of 1 μg/mL, actinomycin D per se induced massive death within 24 hours (Additional Figure 1A) and total cell loss after 2 further days (data not shown). At the lowest and suboptimal concentration tested of 10 ng/mL, the spikes and short neurites that were visible after 2 days of actinomycin D treatment (Additional Figure 1B) remained quite stable for 2 additional days (Additional Figure 1C), while they retracted in 3 more days, accompanied by total cell death (data not shown). In the presence of cycloheximide at the highest tested concentration of 5 μg/mL, some cells elongated short- and medium-length spikes whitin 24 hours (Additional Figure 1D), but the cells remained viable only for 2–3 further days (data not shown). When tested at suboptimal 1 ng/mL concentration, cycloheximide induced a mild neuritogenic effect after 2 days of treatment (Additional Figure 1E), but a clear increase of cell density was visible after about 2 further days (Additional Figure 1F). Finally, we investigated drugs interferingwith microtubule stability. As expected, colchicine preventing microtubule assembly (used at 1 ng/mL), taxol stabilizing microtubules and reducing their dynamicity (used at 10-1000 nM), and vinblastine destabilizing microtubules at minus ends while stabilizing microtubules at plus ends (used at 1 nM), all prevented or inhibited neuritogenesis after 2–3 days (data not shown).
Figure 2 | Ara-C sustains a concentration-dependent and long-lasting neuronal phenotype in NSC-34 cells.
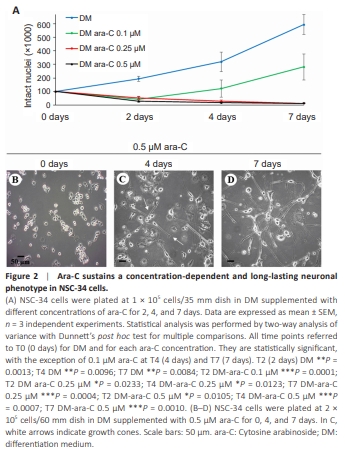
To circumvent the long-term toxic effects of RA or the transient induction of neuronal differentiation by cAMP agents, we hypothesized that the inhibition of cell duplication per se could potentiate the frail neuronal differentiation from NSC-34 cells. The antimetabolite ara-C remains one of the most effective antiproliferative drugs used with duplicating cells. Its actions depend on the conversion to the triphosphate derivative ara-CTP, which is capable of interfering with DNA polymerases and is incorporated into elongating DNA strands, thus leading to DNA fragmentation, duplication chain termination, and eventually apoptosis when a threshold level of DNA damage is exceeded. The concentration of 0.1 μM ara-C for 2 days (Figure 2A) significantly inhibited NSC-34 cell duplication compared with the control DM condition. However, the number of cells in culture progressively increased thereafter, indicating that 0.1 μM ara-C sustains only a transient inhibition of cellproliferation. When ara-C was instead used at 0.25 or 0.5 μM, the antimitotic effect appeared to be highly stable (Figure 2A). Despite a large amount of cell death in DM alone at 24–96 hours after plating (Matusica et al., 2008), the cells roughly duplicated (~2 × 105 ) in DM alone after 2 days in culture, while in the presence of ara-C they were reduced by at least half (less than 5 × 104 ) with respect to the time = 0, thus suggesting that ara-C does not attenuate cell death. However, ara-C at the dose of 0.5 μM prompted a sturdy neuritic arborization that gradually improved with time in culture. In particular, after 4 days the cells elongated more abundant and robust neurites (Figure 2C) compared with control 0 day (Figure 2B), and the neurites became a branching network after 7 days (Figure 2D). When ara-C was used at a concentration of 1 μM instead, it showed some levels of toxicity starting after 7–10 days and thereafter (data not shown).
Figure 3 | Neuronal characterization of ara-C-treated NSC-34 cells.
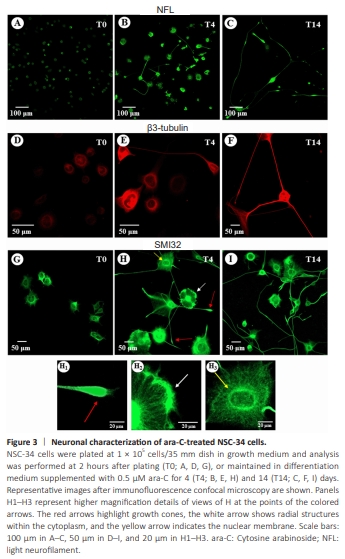
Next, we characterized the biological response of NSC-34 cells to ara-C by performing confocal immunofluorescence analysis of NFL (Figure 3A–C), beta3-tubulin (Figure 3D–F), and nonphosphorylated high neurofilament (SMI32) proteins (Figure 3G–I, H1–H3). Approximately 2 hours after plating and cell adhesion to the substrate (T0), the NFL and SMI32 antibodies stained mainly the cytoplasm and the very short spikes protruded from NSC-34 cells cultured with 0.5 μM ara-C (Figure 3A and G). On day 4 (T4), ara-C prompted a typical neuronal pattern that differed in size, shape, immunofluorescence intensity, and number/length of arborizations per cell (Figure 3B and 3H). In detail, the NFL signal was increased in the enlarged cell bodies and in the protrusions already emerging at this stage (Figure 3B). Moreover, at higher magnification (Figure 3H1–H3), the SMI32 immunopositive staining highlighted different subcellular structures: the red arrows indicate neurites and growth cones present at the edge of the neuritic processes (Figure 3H1); the white arrow highlights radial structures within the cytoplasm, in addition to neurofilament accumulations which are possibly indicative of budding protrusion structures (Figure 3H2); the yellow arrow delineates the nuclear membrane (Figure 3H3). At 14 days (T14, Figure 3I), the majority of the cells possessed long SMI32 bipolar or radial immunopositive neurites. By correlating with the phases of neurogenesis, the marker of neuronal identity beta3-tubulin was highly increased in NSC-34 cells maintained in culture in the presence of 0.5 μM ara-C for 4 days (Figure 3E) compared with T0 (Figure 3D) and it underlined the thick neuritic branches and growth cones at T14 (Figure 3F).
Figure 5 | Long-lasting neurite initiation and regeneration are induced by ara-C.
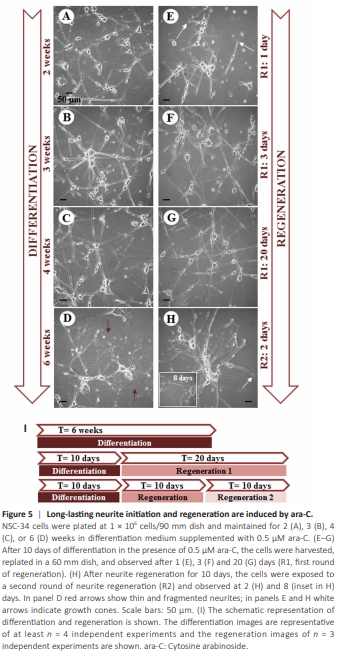
Long-lasting neuronal differentiation is mandatory for legitimating the use of NSC-34 cells as a motor neuron model to dissect neuritogenesis, neurodegenerative mechanisms, and neuroprotective strategies. Thus, we cultured NSC-34 cells in DM supplemented with ara-C for longer time intervals. Notably, the bushy network of highly interconnecting neurite branches that were already evident after ara-C treatment for 7 and 14 days (Figures 2D and 3C, 3F, 3I, respectively) remained consolidated and fully preserved for up to at least 6 weeks in culture (Figure 5A–D). While very long neurites persisted in the majority of the cell population for up to 8 weeks in culture (data not shown), at the latest times shown (6 weeks, Figure 5D) some neurites started appearing thinner and in part fragmented (Figure 5D), some cell bodies initiated shrinking, and cell debris and slight death couldalso be observed as a sign of aging in culture. We next investigated whether the neuronal commitment induced by ara-C could be sustained even after the removal of ara-C. We demonstrated that after 10 days in the continuous presence of 0.5 μM ara-C (but not 0.1 μM, data not shown), NSC-34 cells could be switched to DM without ara-C and they did not duplicate in culture for at least 15 additional days, as directly quantified by counting intact viable nuclei (Additional Figure 2A) and, importantly, they still preserved the neuron-like phenotype (Figure 5B), for up to at least 30 days in culture (data not shown). In other words, they became “neuronal primed” by eliciting a stable neuronal phenotype. Moreover, if switched to GM (without ara-C) some cells visibly escaped from the mitotic arrest and triplicated in about 15 further days (Additional Figure 2A), while others still preserved a neuronal phenotype (Additional Figure 2B). This finding suggests that once differentiated, the cells maintain a post mitotic trait and/or a silent staminality only in the absence of serum factors. The neuritogenic action of 0.5 μM ara-C was remarkably maintained also in DM completely without serum for at least 15 days (data not shown). The neurite regeneration paradigm Having established that NSC-34 cells become “neuronal primed” by ara-C, we next investigated the paradigm of neurite regeneration, a reparative process not requiring new RNA synthesis. In detail (Figure 5I), ara-C fully differentiated cells (0.5 μM for 10 days) were detached from the culture plate and the neurites dismantled. When replated even in the absence of ara-C, the cells adhered to the substrate in 2 hours, and appeared healthy and in good condition (data not shown). On day 1 (Figure 5E), the cells regrew novel processes of variable length and thickness, ending with several visible growth cones (arrows). On day 3 (Figure 5F), the cells underwent full neurite regeneration that persisted for at least 20 days (Figure 5G) in the absence of ara-C. Moreover, regenerated neuronal processes were morphologically indistinguishable from ex novo neurites grown in culture for 3–4 weeks (compare Figure 5G with Figure 5B and C). Remarkably, the neurite regeneration process could be repeated and sustained at least twice in a row (Figure 5H) and the neurites persisted for at least 8 further days in the complete absence of ara-C (inset in Figure 5H). These findings demonstrate that ara-C-primed NSC-34 cells mechanically deprived of neurites remain capable of autonomously reshaping and maintaining their full neuritic network.
Figure 6 | Ki67 proliferation marker is down regulated by treatment with ara-C.
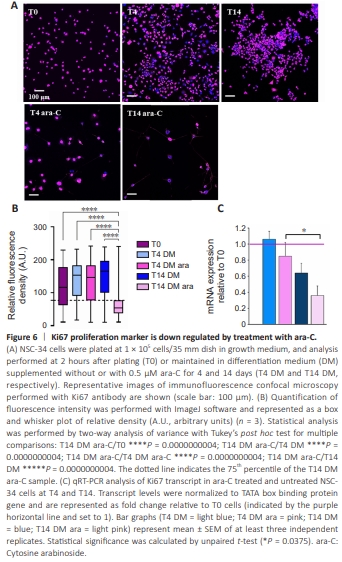
Figure 7 | Neuronal and motor neuronal markers are induced by ara-C.
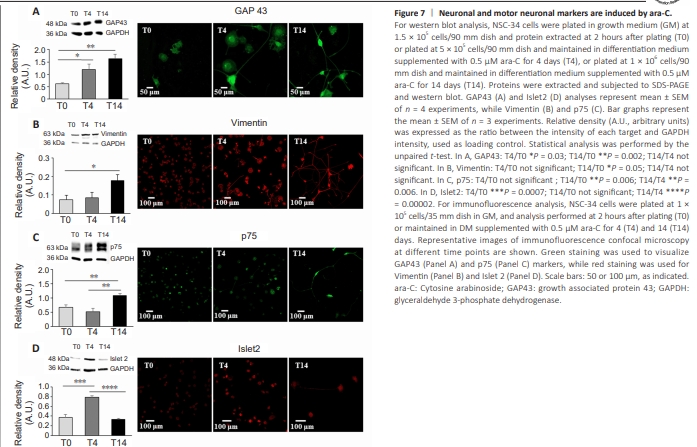
To better characterize ara-C-differentiated NSC-34 cells, we performed qPCR, western blot, and confocal immunofluorescence analysis, using primers and antibodies that recognize markers of either duplication, pan-neuronal, or motor neuronal commitment. We demonstrated that the validated nuclear marker of proliferating tumor cells responsible for heterochromatin organization (Scholzen and Gerdes, 2000; Sobecki et al., 2016), Ki67, significantly and progressively decreased at T4 and T14 after treatment with 0.5 μM ara-C, as shown by immunofluorescence and qPCR analysis (Figure 6). Moreover, the intensities of the neuronal proteins growth associated protein 43 (GAP43) and vimentin increased, as demonstrated by both western blot and confocal analysis (Figure 7A and B). More specific early motor neuron markers such as p75 and Islet2 were also significantly modulated by treatment with ara-C (Figure 7C and D). In detail, the low-affinity p75-NGF receptor is a protein exclusively expressed by motor neurons in the spinal cord, also used to isolate embryonic motor neurons from the lumbar spinal cord (Wiese et al., 2010). NSC-34 cells fully differentiated by ara-C for 14 days displayed increased immunoreactivity for p75 protein (Figure 7C). The transcription factor involved in neuronal fate Islet2 (Tsuchida et al., 1994) was instead significantly but transiently upregulated after 4 days in the presence of ara-C (Figure 7D). The transcription factor Hb9, described as a spinal cord motor neuron-specific marker and critical protein for postmitotic specification, was also transiently induced by ara-C after western blot analysis (Additional Figure 3A). Finally, the immunofluorescence intensity of the choline acetyltransferase enzyme (ChAT), which is responsible for acetylcholine biosynthesis in motor neurons, was highly increased by ara-C after 4 days of treatment, while both cell bodies and abundant neuritic branches were strongly delineated after 14 days (Additional Figure 3B and D). The same occured for the neuron-specific cytoskeletal protein MAP2 enriched in dendrites and perikarya (Additional Figure 3C and D).