脑损伤
-
Figure 3 | C15 induces ChemR23 internalization and regulates the AMPK/NF-κB signaling pathway.
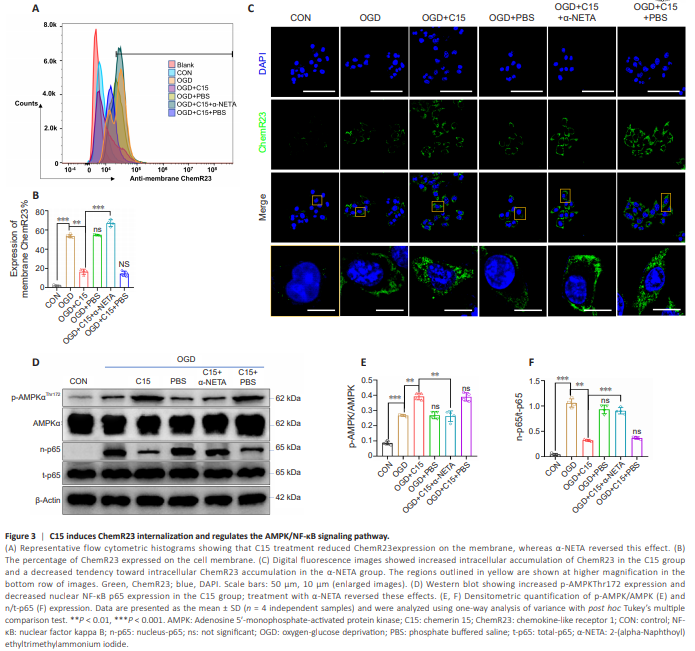
To test this, we treated BV2 cells with C15 and α-NETA (a competitive inhibitor of ChemR23) prior to subjecting the cells to OGD and quantified membrane ChemR23 expression by cytometry and intercellular ChemR23 expression by immunofluorescence staining. In vitro flow cytometry (Figure 3A, B, and Additional Figure 4) showed that 1.56% ± 0.05% of ChemR23 was expressed on the membrane surface in the CON group. OGD increased ChemR23 expression on the surface in BV2 cells compared to the CON group, while treatment with C15 decreased cell-surface ChemR23 expression (54.18% ± 1.63% with OGD alone vs. 18.05% ± 0.88% with OGD + C15). Pretreating BV2 cells with α-NETA increased the percentage of ChemR23 expressed on the membrane surface to 68.68% ± 0.88%. Immunofluorescence staining (Figure 3C) showed that ChemR23 expression was markedly upregulated in the OGDgroup compared with the CON group, and ChemR23 was mainly located on the cell membrane rather than in the cytoplasm, after OGD. C15 treatment increased the intracellular accumulation of ChemR23 compared with the OGD group. As expected, α-NETA counteracted the effect of C15 on ChemR23 internalization. We next investigated downstream signaling events after ChemR23 internalization. ChemR23 can continue signaling after internalization, together with its agonists, and trigger specific downstream effects (Wang et al., 2018a). Chemerin suppresses neuroinflammation by upregulating the ChemR23/AMPK signaling pathway (Zhang et al., 2018). Phosphorylated (active) AMPK helps inhibit NF-κB nuclear translocation (Salminen et al., 2011), thereby reducing the expression of inflammatory cytokines and protecting the brain from I/R injury. To gain mechanistic insights into the anti-inflammatory effects of C15 on BV2 cells, we analyzed the protein expression levels of components of the ChemR23/AMPK/NF-κB signaling pathway. Western blot analysis (Figure 3D– F) revealed that OGD induced the upregulation of p-AMPKThr172 and nuclear NF-κB p65 (n-p65), while total AMPK and NF-κB p65 (t-p65) expression levels remained relatively unchanged among the groups. In contrast, the increase in p-AMPKThr172 was more obvious following C15 administration, and the n-p65 level was reduced, suggesting that C15 promotes AMPK activation and reduces NF-κB p65 translocation into the nucleus through ChemR23. Treatment with α-NETA weakened this effect. In summary, ischemia-hypoxia stimulation induced ChemR23 expression in BV2 cells, and treatment with C15 triggered ChemR23 internalization, thereby regulating the AMPK/NF-κB signaling pathway.
Figure 5 | C15 decreases infarct volume and neuronal apoptosis via ChemR23 in vivo.
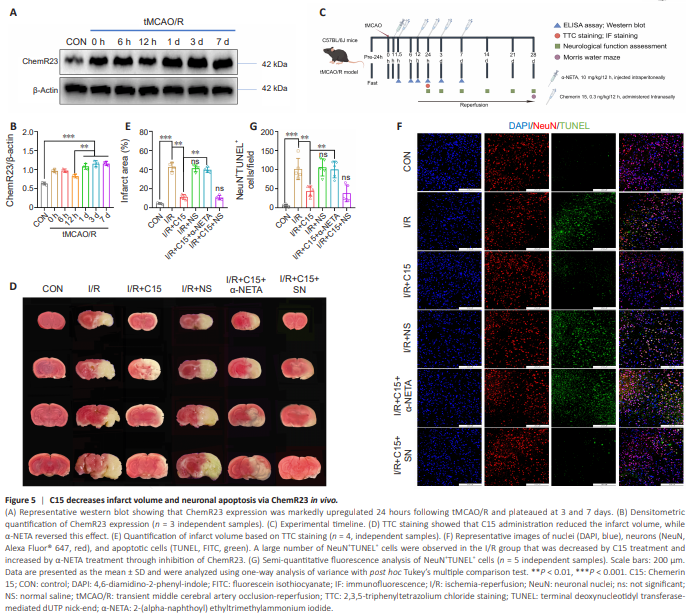
Considering the in vitro results described above, we investigated the antiinflammatory and neuroprotective effects of C15 on mice subjected to I/R injury. To do this, we first assessed ChemR23 expression by western blot at 0, 6, and 12 hours and 1, 3, and 7 days following tMCAO/R. ChemR23 expression increased until 1 day after tMCAO/R, after which it plateaued (Figure 5A and B). Therefore, we investigated the protective effects of C15 on I/R injury and its interaction with ChemR23 at 24 hours post-injury. Before subjecting the mice to tMCAO/R, they were treated with C15 intranasally, as shown in Figure 5C. We assessed the impact of C15 on infarct volume via 2,3,5-triphenyltetrazolium chloride staining. As shown in Figure 5D and E, the percentage of infarct volume out of the total brain volume was 42.76% ± 4.78% in the I/R group, confirming successful establishment of the tMCAO/R model. C15 treatment markedly decreased this percentage to 11.45% ± 2.53%, while α-NETA increased the percentage of infarct volume to 39.74% ± 2.80%. We further explored the effect of C15 on neuronal apoptosis via in vivo double immunofluorescence staining. As shown in Figure 5F and G, the number of apoptotic neurons (NeuN+ TUNEL+ cells) in the I/R group was significantly higher than that seen in the CON group, while the number of apoptotic neurons in the I/R + C15 group was decreased compared with the I/R group. The protective effect of C15 was weakened by treatment with α-NETA. No significant difference was observed in the number of apoptotic neurons between the I/R and I/R + C15 + α-NETA groups.
Figure 6 | C15 regulates microglial phenotype through ChemR23 in vivo.

Subsequently, we conducted immunofluorescence staining to assess microglial phenotypes. As shown in Figure 6A and B, normal brain tissue was almost devoid of IBA1+ cells (activated microglia). The number of IBA1+ cells in the peri-infarct area was increased in the I/R group, demonstrating activation of resting microglia following I/R injury. A large number of IBA1+ CD206+ cells (M2 microglia) and a few IBA1+ CD86+ cells (M1 microglia) were observed in the peri-infarct area in the I/R + C15 group. Intraperitoneal injection of α-NETA led to a remarkable decrease in the number of IBA1+ CD206+ cells and an increase in the number of IBA1+ CD86+ cells. These results imply that C15 reduces infarct volume and neuronal apoptosis and modulates microglial polarization through ChemR23 in mice subjected to I/R injury.