脑损伤
-
Figure 3 | Microglial activation in response to LPS and DEX treatment.
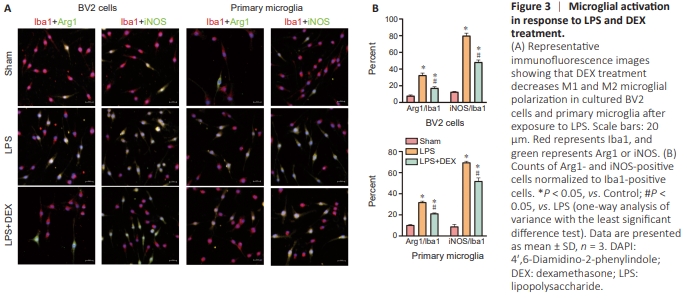
To investigate whether DEX affects polarization of microglia, we used LPS to activate BV2 cells and primary microglia. As expected, BV2 and primary cells expressed both microglial polarization phenotypes (Arg1 and iNOS) after LPS treatment. As a marker of the proinflammatory M1 state, iNOS, was increased by LPS but decreased by DEX treatment. Similarly, DEX also inhibited the LPS-induced elevation in Arg1 expression (Figure 3A). Therefore, microglial stimulation by LPS led to significantly increased expression of both MI and M2 phenotypes (F(2,6) = 129.28, P < 0.05 for BV2-M1, F(2,6) = 611.33.28, P < 0.05 for primary microglia-M1; F(2,6) = 42.77, P < 0.05 for BV2-M2, F(2,6) = 633.8, P < 0.05 for primary microglia-M2), while DEX partially prevented this increase, especially in M1 microglia (P < 0.05 for BV2 and primary microglia; Figure 3B).
Figure 5 | Expression patterns of Arg1 following TBI.
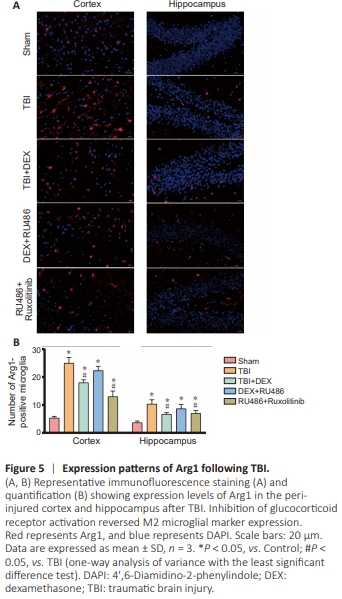
o further investigate the upstream mechanism of microglial polarization of DEX in vivo, we performed qRT-PCR and western blot analysis to measure Arg1, JAK1, and STAT3 levels in the peri-injured cortex and hippocampus after TBI. Neurotrauma significantly enhanced mRNA (F(4,10) = 26.17, P < 0.05 for Arg1; F(4,10) = 30.80, P < 0.05 for JAK1; F(4,10) = 88.16, P < 0.05 for STAT3) and protein levels (F(4,10) = 550.85, P < 0.05 for Arg1; F(4,10) = 736.68, P < 0.05 for JAK1; F(4,10) = 662.32, P < 0.05 for STAT3) of Arg1, JAK1, and STAT3. Compared with the TBI group, DEX significantly reduced their mRNA (P < 0.05) and protein levels (P < 0.05) in the cortex (Figure 4A– C). These trends in gene expression (F(4,10) = 304.91, P < 0.05 for Arg1; F(4,10) = 8.44, P < 0.05 for JAK1; F(4,10) = 31.50, P < 0.05 for STAT3) and their corresponding proteins (F(4,10) = 116.2, P < 0.05 for Arg1; F(4,10) = 112.45, P < 0.05 for JAK1; F(4,10) = 621.49, P < 0.05 for STAT3) were similar in hippocampal tissue. As previously reported, treatment with RU486 (a GR antagonist) inhibited DEX activation of nuclear GR (F(4,10) = 110.4, P < 0.05 in cortex; F(4,10) = 53.67, P < 0.05 in hippocampus). Further, RU486 reversed the effect of DEX on the reduction in Arg1 expression (P < 0.05). In addition, treatment with RU486 and ruxolitinib (an antagonist of JAK1) reduced Arg1 expression compared with the TBI group (P < 0.05). To further investigate the number of M2 microglia, we used immunofluorescence staining. The number of Arg1-positive microglia was significantly decreased in the cortex of the DEX group (F(4,10) = 78.81, P < 0.05 for Arg1) and increased after inhibition of GR activation (P < 0.05). However, there was no statistically significant difference in the number of hippocampal M2 microglia before and after inhibiting GR (F(4,10) = 14.66, P > 0.05 for Arg1) following TBI (Figure 5A and B). These results indicate that DEX inhibited M2 microglial polarization by regulating the GR/JAK1-related pathway after TBI
Figure 6 | RU486 and ruxolitinib treatment decreases DEXinduced apoptosis following TBI.
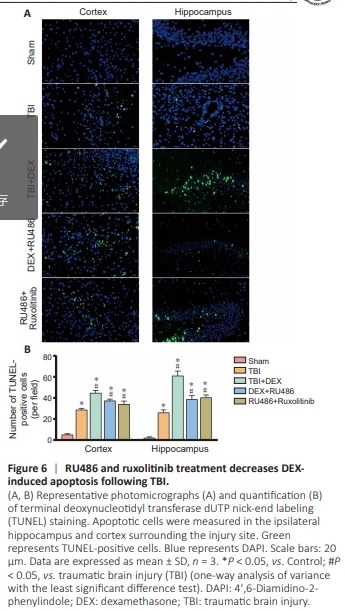
Figure 7 | Inhibition of GR attenuates neuronal death.
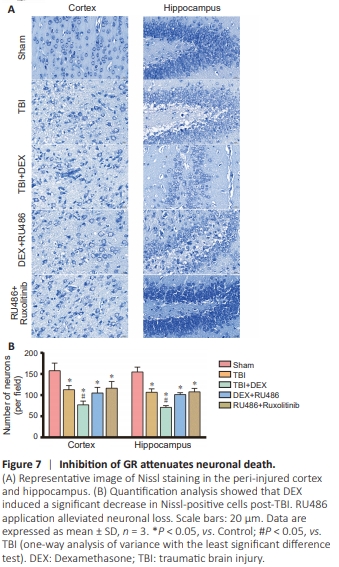
For in vivo studies, we also observed typical apoptosis in the cortex and ipsilateral hippocampus by TUNEL assay (Figure 6A). TUNEL-positive cells were scarcely detectable in Control mice, while the number of apoptotic cells was significantly increased after TBI (F(4,10) = 162.79, P < 0.05 in the cortex; F(4,10) = 147.75, P < 0.05 in the hippocampus) (Figure 6B).DEX treatment enhanced the apoptosis levels after TBI (P < 0.05). However, inhibition of GR reduced the number of TUNEL-positive cells in the cortex and hippocampus (P < 0.05). To further investigate neuronal damage following TBI and DEX treatment, we used Nissl staining. TBI induced a significant decrease in the quantity of Nissl-positive neurons (F(4,10) = 14.21, P < 0.05 in cortex; F(4,10) = 50.69, P < 0.05 in the hippocampus) (Figure 7A and B). Moreover, DEX aggravated neuronal injury following TBI (P < 0.05). After RU486 administration, less neuronal loss was detected compared with the DEX group (P < 0.05). These results indicate that inhibition of GR alleviated neuronal death following DEX treatment after TBI. In addition to participating in apoptosis and neuronal death, GR also influences the structure and function of dendritic spines. Golgi staining revealed a significant decrease in the number of dendritic spines in the TBI group compared with Control mice (F(4,10) = 33.67, P < 0.05 in cortex; F(4,10) = 36.5, P < 0.05 in hippocampus). DEX treatment exacerbated the loss of dendritic spines (P < 0.05), while RU486 mitigated the effect of DEX (P < 0.05) (Figure 8A and B). Together, our neuronal morphology data suggest that DEX altered the complexity of neuronal processes by activating GR. Inhibiting GR can affect the number of dendritic spines, with the possible mechanism being activation of the JAK1/STAT3 signaling pathway.