脑损伤
-
Figure 1 | Accumulation of p-α-syn around the cerebral infarct area in mice subjected to dMCAO.
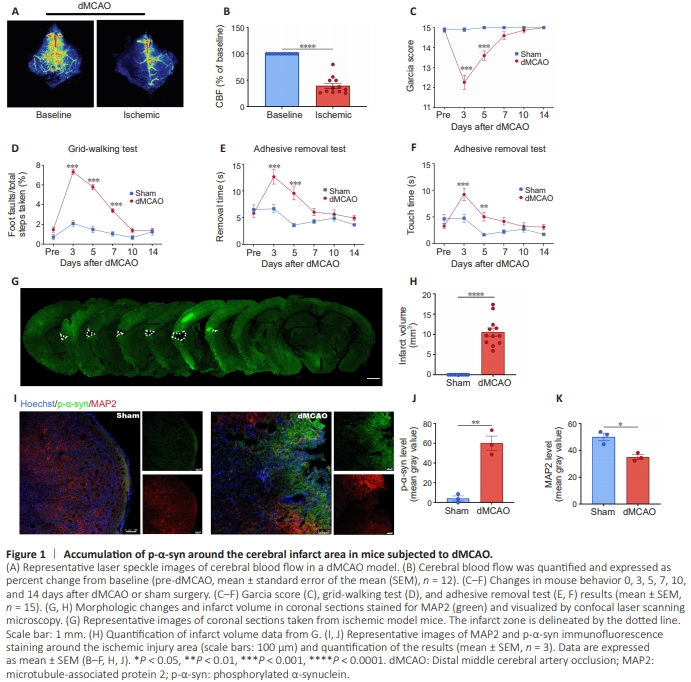
We established a mouse model of IS by performing the classic dMCAO surgery and detected cerebral blood flow by laser speckle imaging. Cerebral blood flow on the infarct side of mice in the dMCAO group was significantly reduced (P < 0.0001) compared with that on the opposite side (Figure 1A and B). Garcia score, grid-walking test, and adhesive removal test were used to evaluate neurological function. The Garcia scores in the dMCAO group were decreased (P < 0.0001) compared with the sham group, indicating that dMCAO induced neurological dysfunction (Figure 1C). The gridwalking test results showed that the foot fault rate of mice in the dMCAO group was significantly greater (P < 0.0001) than that in the sham group, indicating that dMCAO decreased motor coordination (Figure 1D). The time spent touching and removing the adhesive paper in the adhesive removal test was significantly greater in the dMCAO group (P = 0.0002) than in the sham group, indicating that dMCAO impaired sensorimotor function (Figure 1E and F). Taken together, the results from these three tests suggest that the dMCAO mice showed significant neurological dysfunction disorders compared with the sham group. Staining mouse brain sections with the neuronal marker MAP2 showed significant infarct areas in the dMCAO group (Figure 1G), and calculations based on color rendering of the MAP2 staining results demonstrated that the average infarct volume was 10.56 mm3 (Figure 1H). These results indicate that dMCAO caused neurological damage and cerebral infarction in mice. Next, immunofluorescence staining of brain sections from mice in the sham and dMCAO groups was performed to detect p-α-syn. p-α-Syn levels were significantly increased (P = 0.0018) around the infarct area in dMCAO mice, and more p-α-syn aggregation was observed in dMCAO mice, compared with the sham mice, while MAP2 immunoreactivity was significantly decreased (P = 0.0103) in dMCAO mice compared with sham mice (Figure 1I–K). These findings suggest that periinfarct nerve injury was associated with pathological p-αsyn deposition.
Figure 2|Accumulation of p-α-syn in the brain of hypoxic model mice.
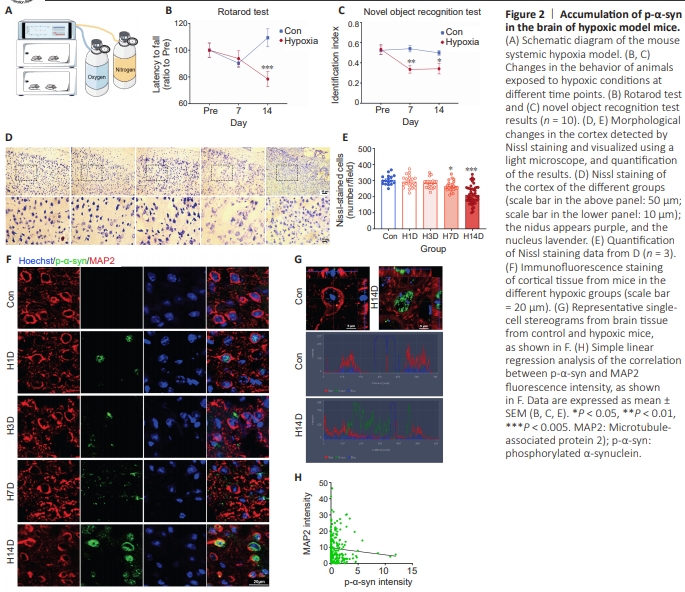
The pathological changes caused by ischemia are mainly due to the reduced use of oxygen in the body and the formation of a hypoxic internal environment (Eltzschig and Eckle, 2011; Taylor and Colgan, 2017). Therefore, we speculated that hypoxia caused the observed increase in p-α-syn expression and aggregation. To test this, we constructed a whole-body hypoxic mouse model by housing mice in a hypoxic chamber with 13% O2 for 2 weeks (Figure 2A) and then performed the rotarod test and novel object recognition test to assess cognition and motor function, respectively. Compared with the control group, 7 days of persistent hypoxia resulted in cognitive impairment (P < 0.01), and 14 days of persistent hypoxia further led to significant impairment (P < 0.001) of motor function (Figure 2B and C). Next, Nissl staining of brain tissue slices from the different groups of mice was performed to detect neuronal damage. After 7 days of hypoxia, the Nissl particles were blurred, the margins were not clear, and the number of neurons was significantly decreased (P < 0.05) compared with the control group (Figure 2D and E). Furthermore, immunofluorescence staining showed that hypoxia induced a significant increase in p-α-syn levels in the cortical neurons compared with the control group. p-αSyn gradually accumulated in the neurons as the duration of exposure to hypoxic conditions increased (Figure 2F and G), and the p-α-syn fluorescence intensity was negatively correlated with the MAP2 fluorescence intensity (Figure 2H). These results suggest that hypoxia induced α-syn phosphorylation and aggregation in the brain, which may be an important cause of hypoxia-induced neuronal injury