脑损伤
-
Figure 3 | CARD19 expression is increased in microglia after ischemic stroke.
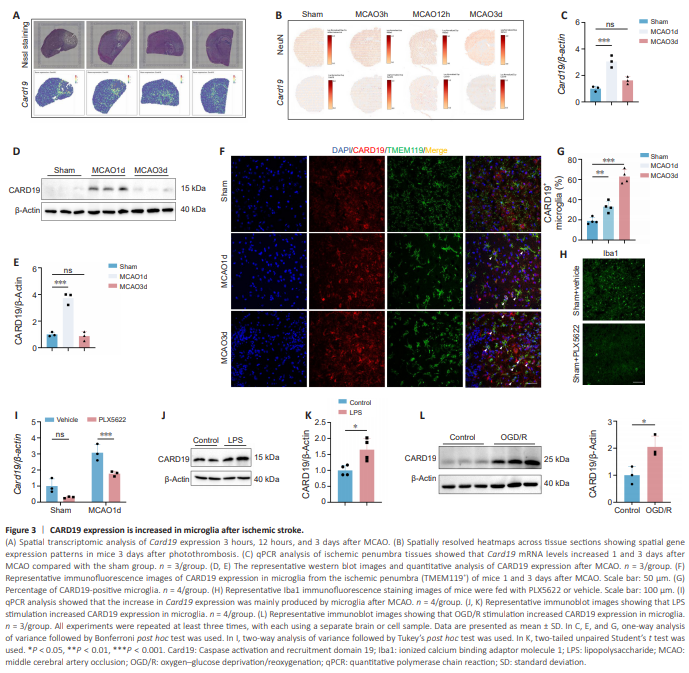
We and others have recently performed spatial transcriptomic sequencing of ischemic brain tissue collected from MCAO mice (Li et al., 2023a; Han et al., 2024). After reanalyzing the published data, we found that Card19 expression was markedly increased in microglia 3 hours and 12 hours after MCAO compared with the sham group. Notably, the spatial transcriptomic results showed that the elevated Card19 expression was mainly distributed in the ischemic penumbra (Figure 3A and B). qPCR and western blotting confirmed that Card19 mRNA and protein levels were significantly increased in the ischemic penumbra at 1 and 3 days after MCAO (Figure 3C–E). Furthermore, immunofluorescence staining showed that compared with the sham group, microglial CARD19 expression was increased 1 and 3 days after MCAO (Figure 3F and G). To determine whether microglia were responsible for the elevated Card19 expression levels, mice were fed 8 g feed containing PLX5622 (a CSF1R inhibitor) daily for 2 weeks to deplete microglia. The efficiency of microglial depletion was verified by immunofluorescence staining (Figure 3H). The results showed that, the elevated Card19 mRNA levels observed after MCAO decreased significantly to physiological levels after microglia depletion (MCAO1d + Vehicle vs. MCAO1d + PLX5622, P = 0.0081), indicating that the increased CARD19 expression seen after MCAO was mainly derived from microglia (Figure 3I). Since DAMPs can act on microglial Toll-like receptors to initiate a proinflammatory response after ischemic stroke, we stimulated cells with LPS in vitro to mimic microglial activation during the acute phase of ischemic stroke. The results showed that treating primary microglia with LPS increased CARD19 expression compared with the control group (Figure 3J and K). Moreover, when oxygen–glucose deprivation/reoxygenation (OGD/R) was applied to microglia to mimic oxygen and energy deficiency after ischemic stroke, CARD19e protein levels increased compared with the control group (Figure 3L). Taken together, these findings suggest that microglial CARD19 expression increases in the penumbra after ischemic stroke.
Figure 4 | Microglial CARD19 deficiency aggravated ischemic brain injury.
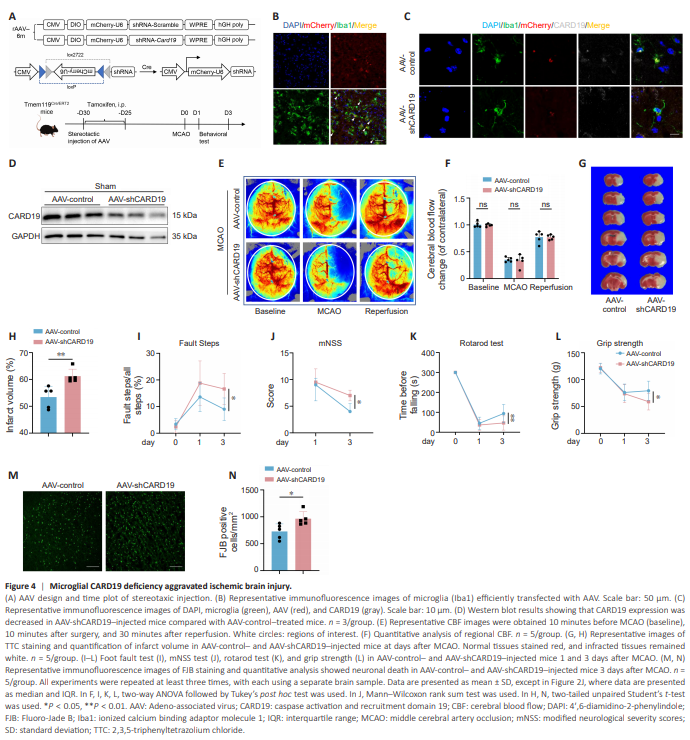
To investigate the role of microglial CARD19 in ischemic stroke, we conditionally knocked down Card19 expression in microglia through stereotaxic injection of AAVs into the cerebral cortex of Tmem119Cre/ERT2 mice (Figure 4A). Immunofluorescence staining showed that microglia were efficiently transfected, and CARD19 knockdown was successful (Figure 4B– D and Additional Figure 1D–F). Co-staining for AAV and other cell types showed that the majority of AAV co-localized with microglia, while only a minimal amount of AAV was detected in astrocytes. There was no significant co-localization of AAV with endothelial cells (CD31) or neurons (NeuN) (Additional Figure 1A–C). These findings suggested that AAV transfected microglia and specifically knocked down CARD19. Next, mice in the AAVcontrol group and AAV-shCARD19 group were subjected to MCAO. There are no significant differences in regional CBF between AAV-control and AAVshCARD19 mice at minutes before MCAO (baseline), 10 minutes after surgery, or 30 minutes after reperfusion (Figure 4E and F). TTC staining showed that the infarct volume of AAV-shCARD19 mice was larger than that of AAV-control mice (Figure 4G and H). A series of behavioral tests (foot fault test, mNSS test, rotarod test, and grip strength test) showed that the AAV-shCARD19 mice exhibited more severe neurological deficits than the AAV-control mice (Figure 4I–L). Fluoro-Jade B staining showed that more neurons died in AAVshCARD19 mice than in AAV-control mice (Figure 4M and N). These results suggested that conditional knockdown of CARD19 in the microglia aggravates brain injury after ischemic stroke.
Figure 5 | Microglial CARD19 deficiency amplifies post-stroke neuroinflammatio.
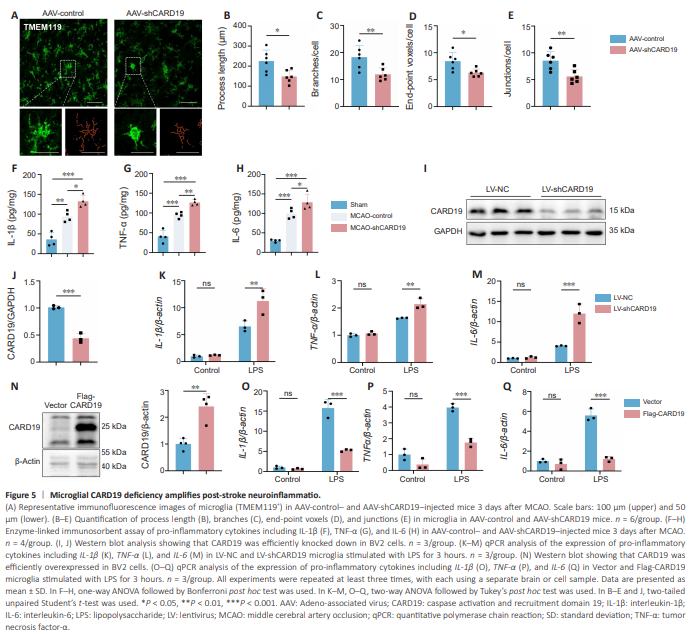
Since microglia plays a pro-inflammatory role during the acute phase of ischemic stroke, we next asked whether microglial CARD19 deficiency affects neuroinflammation post-stroke. Homeostatic microglia have small cell bodies and ramified branches. After ischemic stroke, microglia are activated rapidly and adopt an amoeboid-like phenotype (Jia et al., 2021). We therefore observed microglial morphology in the ischemic penumbra by TMEM119 immunofluorescence staining. Microglia in the AAV-shCARD19 mice exhibited larger cell bodies and shorter and less ramified branches than microglia in the AAV-control mice on the 3rd day after MCAO (AAV-control vs. AAV-shCARD19: process length, P = 0.0150; branches, P = 0.0099; end-point voxels, P = 0.0129; junctions, P = 0.0060; Figure 5A–E). Furthermore, compared with AAVcontrol mice, AAV-shCARD19 mice exhibited higher levels of pro-inflammatory factors, including IL-1β, TNF-α, and IL-6 (IL-1β: Sham vs. MCAO-control, P = 0.0011; Sham vs. MCAO-shCARD19, P < 0.0001; MCAO-control vs. MCAOshCARD19, P = 0.0142; TNF-α: Sham vs. MCAO-control, P = 0.0002; Sham vs. MCAO-shCARD19, P < 0.0001; MCAO-control vs. MCAO-shCARD19, P = 0.0066; IL-6: Sham vs. MCAO-control, P = 0.0002; Sham vs. MCAO-shCARD19, P < 0.0001; MCAO-control vs. MCAO-shCARD19, P = 0.0435; Figure 5F–H). To further investigate the role of CARD19 in microglia, we knocked down or overexpressed CARD19 in BV2 cells using lentiviruses (Figure 5I and J) and found that the levels of IL-1β, TNF-α, and IL-6 mRNA in LV-shCARD19-treated microglia were significantly higher than those in LV-NC-treated microglia after LPS stimulation (IL-1β/β-actin: LV-NC + LPS vs. LV-shCARD19 + LPS, P = 0.0074; TNF-α/β-actin: LV-NC + LPS vs. LV-shCARD19 + LPS, P = 0.0020; IL-6/β-actin: P = 0.0002; Figure 5K–M). Consistent with this, CARD19 overexpression suppressed IL-1β, TNF-α, and IL-6 mRNA levels after LPS stimulation (Figure 5N–Q). Thus, CARD19 deficiency promotes microglial activation and amplifies neuroinflammation in ischemic stroke.
Figure 6 | Microglial CARD19 deficiency accelerates ultrastructural and functional damage to mitochondria in ischemic stroke.
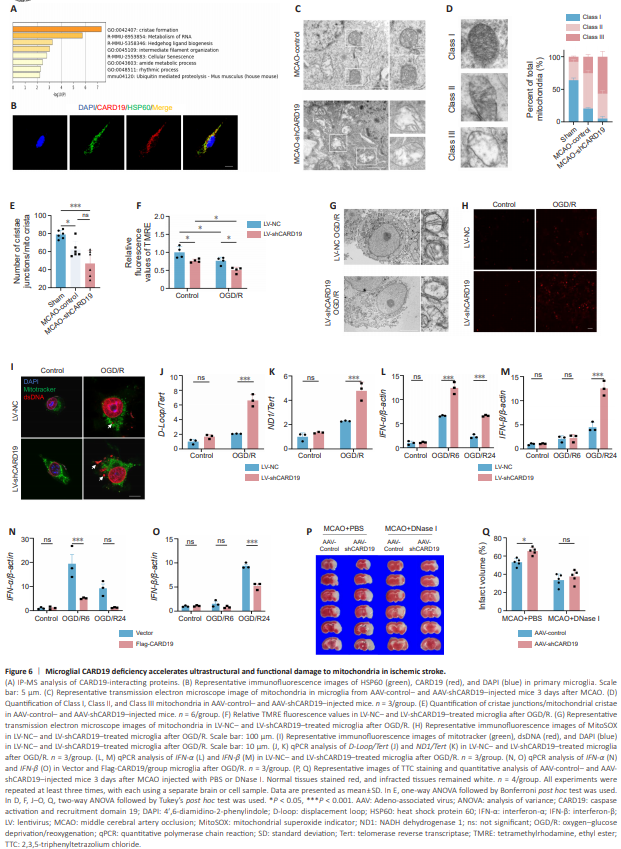
To investigate the mechanism underlying the neuroprotective role of microglial CARD19, we screened for CARD19-interacting proteins by immunoprecipitation-mass spectrometry. The proteins that interacted with CARD19 in microglia were enriched in the mitochondrial cristae formation pathway (Figure 6A). Immunofluorescence staining confirmed that CARD19 co-localized with HSP60 (a marker of mitochondria) in primary microglia (Pearson’s_Rr = 0.84222891), indicating that CARD19 may play a functional role in the mitochondria of microglia (Figure 6B and Additional Figure 2). When ischemic stroke occurs, cerebral blood flow is blocked, resulting in microglial mitochondria dysfunction, which manifests as reduced ATP production, energy metabolism disorders, and mitochondrial structural damage (Li et al., 2014; Yang et al., 2018). To investigate the role of CARD19 in the maintenance of mitochondrial cristae in microglia, the mitochondrial ultrastructure of microglia in the ischemic penumbra on day 3 after MCAO was observed by transmission electron microscopy (Figure 6C). Our results showed that, after MCAO, the proportion of Class I mitochondria was significantly reduced from 60%–70% to 20%, that of Class II mitochondria was significantly increased from 20%–30% to 50%–60%, and that of Class III mitochondria was significantly increased from 10% to 20%–30% compared with sham controls (Sham vs. MCAO-control: Class I, P < 0.0001; Class II, P < 0.0001; Class III: P < 0.0001). However, the proportion of Class I mitochondria was significantly reduced to around 10%, that of Class II mitochondria was reduced 30%–40%, and that of Class III mitochondria was increased to 50%–70% in AAV-shCARD19-treated mice compared with AAV-control-treated MCAO mice (MCAO-control vs. MCAO-shCARD19: Class I, P = 0.0079; Class II, P = 0.0050; Class III: P < 0.0001; Figure 6D). Additionally, after MCAO, the ratio of cristae junctions/mitochondrial cristae was decreased in AAVshCARD19-treated mice compared with AAV-control-treated mice, suggesting an increasing in cristae disintegration (Sham vs. MCAO-control, P = 0.0361; Sham vs. MCAO-shCARD19, P = 0.0002; MCAO-control vs. MCAO-shCARD19, P = 0.0539; Figure 6E). Moreover, the results from electron microscopy analysis of OGD/R-treated microglia cells were consistent with those obtained in mice (Figure 6G). These findings suggest that microglial CARD19 deficiency exacerbates mitochondrial structural damage after ischemic stroke. After ischemic stroke, hypoxia induces decoupling of the mitochondrial respiratory chain, leading to a surge in superoxide ROS production in mitochondria, as well as a decline in the mitochondrial membrane potential (An et al., 2021). Mitochondrial oxidative stress subsequently disrupts mtDNA integrity and promotes the release of oxidized mtDNA into the cytoplasm (Kim et al., 2023). Therefore, we examined relevant indicators of mitochondrial dysfunction. Compared with LV-NC-treated microglia, mitochondrial ROS production and mtDNA release were increased in the LV-shCARD19 group (D-Loop/Tert: LV-NC + OGD/R vs. LV-shCARD19 + OGD/R, P < 0.0001; ND1/ Tert: LV-NC + OGD/R vs. LV-shCARD19 + OGD/R, P = 0.0003; Figure 6H–K). In addition, the mitochondrial membrane potential was decreased in LVshCARD19-treated mice compared with LV-NC-treated microglia (LV-NC + Control vs. LV-shCARD19 + Control, P = 0.0393; LV-NC + Control vs. LVNC + OGD/R, P = 0.0404; LV-shCARD19 + Control vs. LV-shCARD19 + OGD/ R, P = 0.0255; LV-NC + OGD/R vs. LV-shCARD19 + OGD/R, P = 0.0248; Figure 6F). These findings suggest that microglial CARD19 deficiency inhibits mitochondrial function under OGD/R conditions. Previous studies have shown that disruption of mitochondrial cristae leads to mtDNA release and mtDNA-dependent interferon I responses (He et al., 2022). To determine whether microglial CARD19 deficiency–induced mtDNA release activates the IFN-I response, we examined IFN-α and IFN-β mRNA levels by qPCR and found that CARD19 knockdown boosted the OGD/R-induced increase in IFN-α and IFN-β expression (Figure 6L and M). Conversely, CARD19 overexpression abolished the OGD/R-induced IFN-I response (Figure 6N and O). Next, we depleted cytoplasmic mtDNA by intraperitoneal injection of DNase I. TTC staining showed that mtDNA clearance partially reversed the adverse outcomes caused by microglial CARD19 deficiency (Figure 6P and Q). These results suggested that microglial CARD19 deficiency aggravates brain injury by promoting mitochondrial damage and provoking mtDNA-dependent interferon I responses.
Figure 7 | Microglial CARD19 deficiency accelerates apoptosis in ischemic stroke.
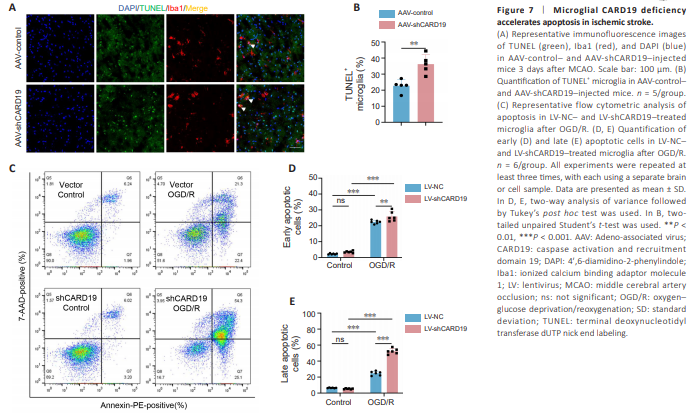
Mitochondrial dysfunction, along with increased ROS production and calcium overload, occur after ischemic stroke, which results in opening of the mitochondrial membrane permeability transition pore, release of cytochrome c, and consequent activation of effector caspases, ultimately leading to apoptosis (Sims and Muyderman, 2010). Thus, we investigated the effect of microglial CARD19 deficiency on apoptosis in vivo. TUNEL staining showed that, compared with AAV-control-treated mice, the proportion of TUNEL+ Iba1+ cells in Iba1+ cells was significantly reduced in AAV-shCARD19-treated mice (Figure 7A and B). Next we detected apoptosis of cultured microglia after OGD/R in both LV-NC- and LV-shCARD19-treated microglia using an Annexin V-PE/7-AAD apoptosis detection kit and found that, compared with LV-NC– treated microglia, the Annexin-V (+) 7-AAD (–) and Annexin-V (+) 7-AAD (+) ratios were significantly increased in LV-shCARD19-treated microglia (early apoptosis: LV-NC + Control vs. LV-NC + OGD/R, P < 0.0001; LV-shCARD19 + Control vs. LV-shCARD19 + OGD/R, P < 0.0001; LV-NC + OGD/R vs. LVshCARD19 + OGD/R, P = 0.0080. Late apoptosis: LV-NC + Control vs. LV-NC + OGD/R, P < 0.0001; LV-shCARD19 + Control vs. LV-shCARD19 + OGD/R, P < 0.0001; LV-NC + OGD/R vs. LV-shCARD19 + OGD/R, P < 0.0001; Figure 7C– E). These findings suggested that CARD19 deficiency aggravates microglial apoptosis after brain ischemia. Therefore, microglial CARD19 deficiency aggravates the destruction of mitochondrial cristae, leading to mitochondrial dysfunction in ischemic stroke.