NRR:广州大学杨莉/华南师范大学龙程课题组揭示淀粉样前体蛋白调控成年新生神经元成熟及存活的新机制
作者:胡海东
审核:杨莉
阿尔茨海默病(Alzheimer‘s disease, AD)是一种以β-淀粉样蛋白(Amyloid-β, Aβ)沉积、 Tau蛋白神经原纤维缠结、神经元丢失以及认知功能障碍为主要特征的神经退行性疾病[1]。Aβ由其前体蛋白——淀粉样前体蛋白(Amyloid Precursor Protein, APP)经β和g分泌酶剪切产生。目前AD研究多聚焦于Aβ的神经毒性及其靶向药物的开发[2-3],尚不明确APP的生理功能及其缺失在AD发病中的作用。
APP作为一种多次跨膜的蛋白,其本身及代谢产物对神经元功能,如离子通道活动、突触传递、神经振荡和认知行为等具有重要的调控作用[4-5]。海马体是学习记忆的关键脑区,也是AD早期易受损的重要脑区[6]。其齿状回(Dentate Gyrus, DG)是大脑中少数终身持续产生新生神经元(Adult-born Granule Cells, abGCs)的区域之一[7-8]。这些abGCs经历增殖、分化、迁移和成熟的过程,最终整合到已有的神经网络中,对海马依赖的学习记忆至关重要[9]。神经元丢失和成年神经发生减弱是AD的关键特征[10]。此前的免疫细胞化学研究表明,APP缺失会增加DG神经前体细胞的发生,但同时降低abGCs的存活率[11]。然而,APP缺失如何影响abGCs在不同成熟阶段的电生理特性、形态特征、分子表达、存活及其背后的分子机制仍有待阐明。
广州大学杨莉教授和华南师范大学龙程教授课题组在《中国神经再生研究(英文)》(《Neural Regeneration Research》)发表的研究利用APP基因敲除(APP-/-)小鼠模型,结合逆转录病毒标记、全细胞膜片钳记录、免疫荧光染色和行为学测试等技术,系统探究了APP缺失对不同周龄(4周和10周)abGCs以及成熟颗粒细胞(Mature Granule Cells, mGCs)的电生理特性、形态成熟、突触功能和细胞存活的影响。研究发现,APP缺失导致10周龄abGCs出现内在兴奋性和微小突触后电流活动异常增高、树突形态发育不良、钾氯协同转运蛋白2(Potassium Chloride Cotransporter 2, KCC2)表达下调。此外,APP缺失鼠在情境条件恐惧(Contextual Fear Conditioning, CFC)测试中表现出冻结行为增加。该研究揭示了APP通过调控KCC2表达维持神经元氯离子稳态和抑制性张力,从而保障abGCs正常成熟和存活的关键作用,为揭示APP在海马神经元新生及功能障碍中的作用提供了新视角。
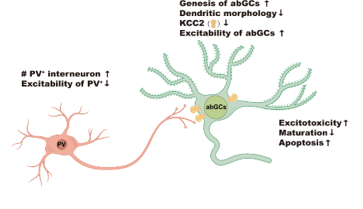
研究者首先通过逆转录病毒注射特异性标记abGCs,发现APP敲除显著增加了注射病毒后4周(4 WPI)和10周(10 WPI)的abGCs数量(图1A,图2A),与之前报道的神经增殖增加一致[11]。
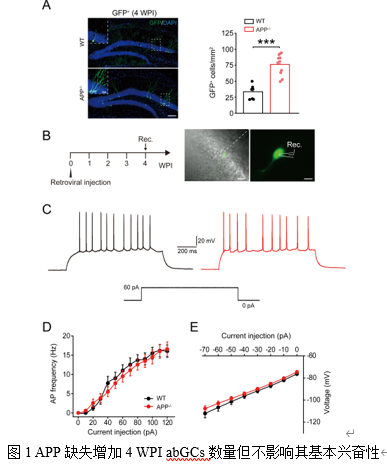
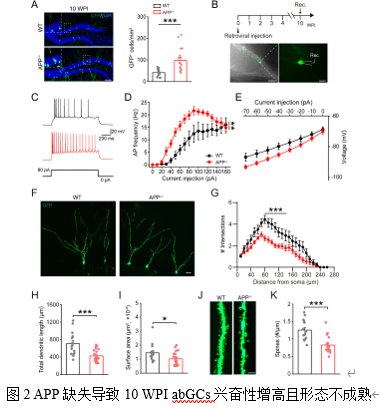
在电生理特性上,4 WPI的abGCs在APP-/-小鼠中动作电位(Action Potential, AP)发放频率与野生型(Wild Type, WT)无差异(图1B-D),但其AP的半幅宽和下降支时间延长(表1),提示其电生理性质已出现不成熟的迹象。相比之下,10 WPI的abGCs在APP-/-小鼠中表现出明显的过度兴奋:其AP发放频率显著增高(图2B-D),起始阈值(Rheobase)降低,输入电阻增高(表2)。同时,这些细胞的树突复杂性降低,总长度和表面积减少,树突棘密度也显著下降(图2F-K)。
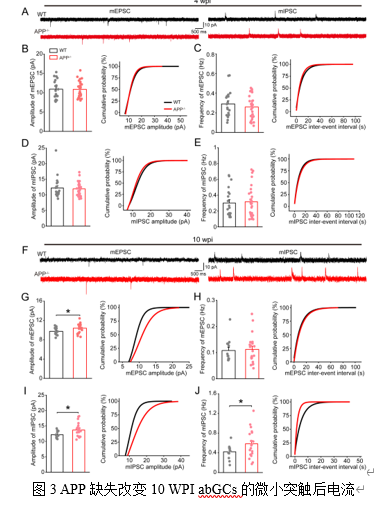
在突触功能方面,4 WPI
abGCs的微小兴奋性/抑制性突触后电流(mEPSC/mIPSC)无显著变化(图3A-E)。然而,10 WPI abGCs的mEPSC振幅和mIPSC的振幅、频率均显著增加(图3F-J),表明兴奋性和抑制性突触传递均增强。
有趣的是,与过度兴奋的10 WPI
abGCs相反,APP-/-小鼠中无法被病毒标记的、更古老的mGCs则表现出兴奋性降低,AP发放频率减少,发放阈值增高(图4A-C,表3)。其mIPSC频率增加,但振幅未变(图4D-H)。
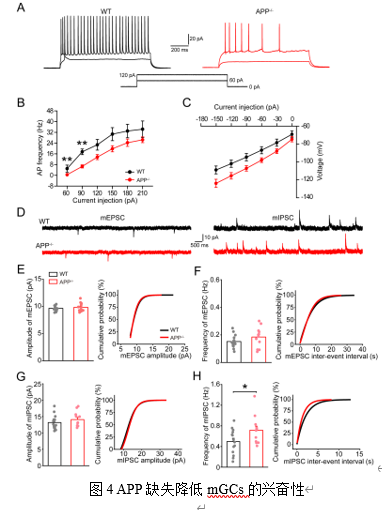
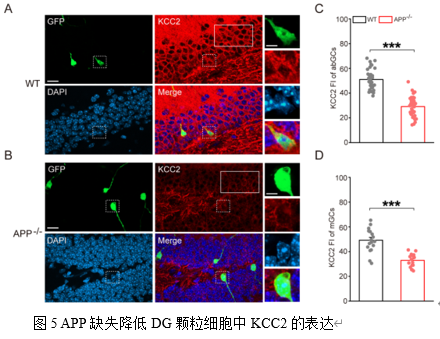
为进一步探究机制,研究者检测了KCC2的表达。KCC2是维持神经元内低氯离子浓度和GABA能抑制性功能的关键蛋白[12]。我们以往在海马CA1的研究表明APP与KCC2间存在着直接作用(Protein-protein Interaction),APP缺失影响KCC2的络氨酸磷酸化致使其更多的降解[5],我们因此探索了DG的KCC2水平。结果显示,APP缺失导致10
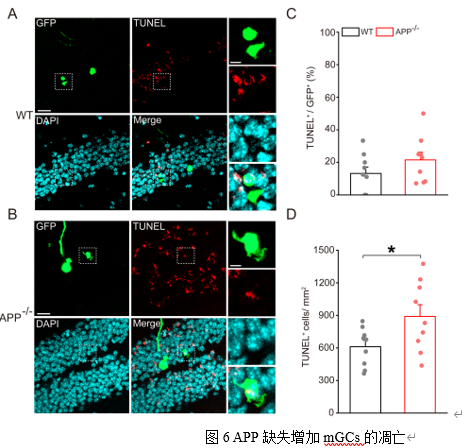
WPI abGCs和mGCs中的KCC2水平均显著下调(图5A-D)。此外,TUNEL凋亡检测发现APP-/-小鼠DG区的mGCs凋亡信号显著增强(图6A-D)。KCC2功能缺失可导致神经元兴奋性毒性和凋亡[13],本研究结果提示了KCC2下降可能介导APP缺失所致的GCs凋亡。
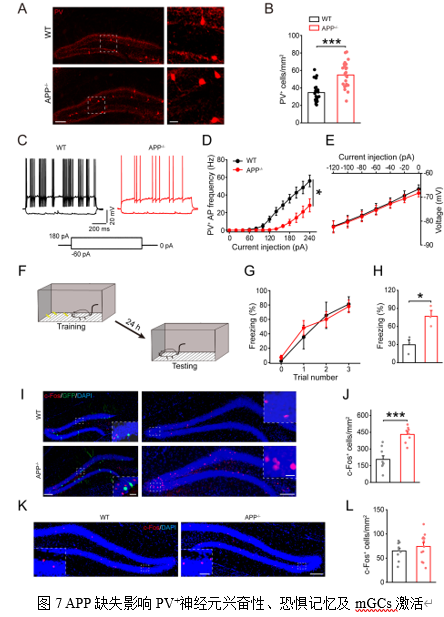
对DG区内主要的GABA能抑制性中间神经元——Parvalbumin阳性(PV+)神经元的分析发现, APP-/-小鼠中PV+神经元的内在兴奋性降低了(图7C-E),这可能削弱了其对颗粒细胞的抑制控制。此外,抑制性受体GABAARα1和α5亚基的水平下降,而BDNF的受体TrkB表达上调,这可能是一种代偿机制。
在行为层面,APP-/-小鼠在情境条件恐惧(Contextual Fear
Conditioning, CFC)测试中表现出冻结行为增加(图7G-H)。进一步检测发现,CFC后APP-/-小鼠DG区被激活的(c-Fos阳性)mGCs数量显著多于WT小鼠(图7I-J),但被病毒标记的abGCs并未被激活(图7I),表明APP缺失导致mGCs过度激活,但abGCs并未参与该恐惧记忆过程。
该研究系统地揭示了APP缺失以细胞年龄依赖的方式影响abGCs的功能成熟和存活:① 使10周龄abGCs停滞在电生理和形态学不成熟的过度兴奋状态;② 下调KCC2表达,改变GABA能抑制功能稳态,可能诱发兴奋性毒性;③ 最终导致成熟的mGCs发生凋亡。abGCs的异常增多可能是一种针对神经元丢失的代偿性反应,但无法挽救整体的网络功能障碍。PV+中间神经元兴奋性降低和GABA受体减少共同导致了抑制性张力削弱,进一步加剧了网络兴奋/抑制(E/I)平衡的失调。APP-/-小鼠表现出的恐惧记忆异常和mGCs过度激活,可能与DG区模式分离功能受损有关。
该研究的不足之处在于:首先,使用全身性APP敲除小鼠无法区分APP功能缺失效应与发育过程中的代偿适应性改变。未来研究利用条件性基因敲除技术,在成年神经发生环境细胞中特异性敲除APP,将有助于阐明其细胞自主与非细胞自主性作用机制。其次,离体脑片的电生理记录可能无法完全模拟在体状态的复杂网络活动。缺乏对单个abGCs的长期追踪记录,也限制了在10 WPI超兴奋性与后续mGCs凋亡之间建立直接的因果联系。
总之,该研究不仅揭示了APP在调控abGCs成熟中的关键作用,其发现的“KCC2表达下降→abGCs过度兴奋→mGCs凋亡”这一病理过程,与AD早期海马神经元过度活跃先于神经元丢失的假设[14]相吻合。AD大脑除了Aβ沉积还有APP功能受损,因此,该研究为AD的发病机制补充了了新的可能,提示靶向干预成年神经发生过程或KCC2功能可能成为AD治疗的潜在新策略。
参考文献#br#
[1] Breijyeh Z, Karaman R (2020) Comprehensive review on Alzheimer's disease: causes and treatment. Molecules 25:5789.#br#
[2] Zhang Y, et al. (2023) Amyloid beta-based therapy for Alzheimer's disease: challenges, successes and future. Signal Transduct Target Ther 8:248.#br#
[3] Vitek GE, et al. (2023) Lecanemab (BAN2401): an anti-beta-amyloid monoclonal antibody for the treatment of Alzheimer disease. Expert Opin Investig Drugs 32:89-94.#br#
[4] Yang L, et al. (2009) Amyloid precursor protein regulates Cav1.2 L-type calcium channel levels and function to influence GABAergic short-term plasticity. J Neurosci 29:15660-15668.#br#
[5] Chen M, et al. (2017) APP modulates KCC2 expression and function in hippocampal GABAergic inhibition. Elife 6:e20142.#br#
[6] Rao YL, et al. (2022) Hippocampus and its involvement in Alzheimer's disease: a review. 3 Biotech 12:55.#br#
[7] Denoth-Lippuner A, Jessberger S (2021) Formation and integration of new neurons in the adult hippocampus. Nat Rev Neurosci 22:223-236.#br#
[8] Gage FH (2025) Adult neurogenesis in the human dentate gyrus. Hippocampus 35:e23655.#br#
[9] Miller SM, Sahay A (2019) Functions of adult-born neurons in hippocampal memory interference and indexing. Nat Neurosci 22:1565-1575.#br#
[10] Salta E, et al. (2023) Adult hippocampal neurogenesis in Alzheimer's disease: A roadmap to clinical relevance. Cell Stem Cell 30:120-136.#br#
[11] Wang B, et al. (2014) The amyloid precursor protein controls adult hippocampal neurogenesis through GABAergic interneurons. J Neurosci 34:13314-13325.#br#
[12] Ben-Ari Y (2002) Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci 3:728-739.#br#
[13] Kontou G, et al. (2021) KCC2 is required for the survival of mature neurons but not for their development. J Biol Chem 296:100364.#br#
[14] Palop JJ, Mucke L (2010) Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci 13:812-818.#br#