中国神经再生研究(英文版) ›› 2026, Vol. 21 ›› Issue (8): 3717-3729.doi: 10.4103/NRR.NRR-D-24-01612
抑制Tp53表达减轻脊髓损伤后的铁死亡
TP53 drives neuronal ferroptosis by promoting KLHL4-mediated SLC7A11 ubiquitination after spinal cord injury
Yu Kang1, 2, 3, #, Qiangwei Li4, #, Tianlun Zhao1, #, Haojie Zhang1, #, Yuejian Sun1, 2, Yilong Zhang1, 2, Da An1, 2, Zongsheng Yin3, *, Yong Xuan3, 5, *, Peigen Xie1, 2, *
- 1Department of Spine Surgery, The Third Affiliated Hospital, Sun Yat-sen University, Guangzhou, Guangdong Province, China;
2Department of Orthopedics, Zhaoqing Hospital, The Third Affiliated Hospital of Sun Yat-sen University, Zhaoqing, Guangdong Province, China;
3Department of Orthopedics, The First Affiliated Hospital of Anhui Medical University, Hefei, Anhui Province, China;
4School of Basic Medical Sciences, Anhui Medical University, Hefei, Anhui Province, China;
5Department of Orthopedics, The Second People’s Hospital of Hefei, Hefei, Anhui Province, China
摘要:
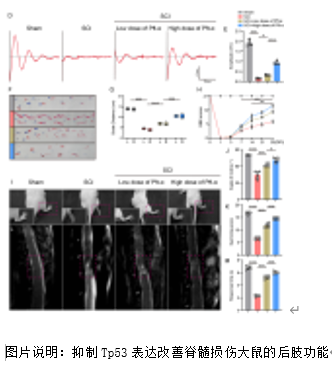
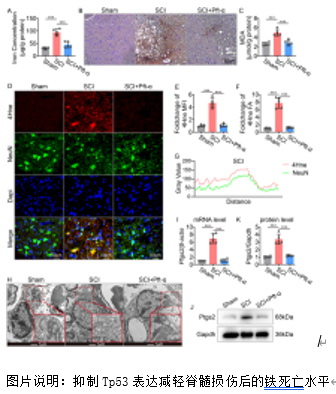
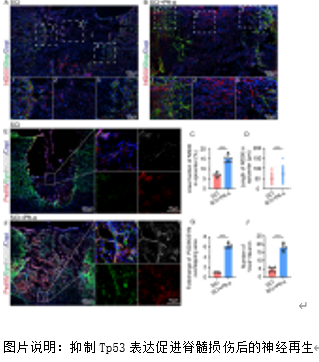
铁死亡是脊髓损伤后发生的关键病理事件,对神经功能的恢复构成重大挑战。Slc7a11在维持细胞氧化还原稳态和抵抗铁死亡方面至关重要。脊髓损伤后Slc7a11下调导致的神经元铁死亡的机制尚不明确。研究提供了证据表明,作为Slc7a11的负调节因子,Tp53在脊髓损伤后显著上调。转录组分析显示Tp53与损伤严重程度相关。随后,实验通过脊髓挫伤大鼠模型验证了脊髓损伤发生后,Tp53 的表达会上升,并在与 mRNA 转录和蛋白质泛素化相关的生物过程中显著富集;抑制 Tp53 显著减轻了脊髓损伤大鼠的铁死亡表型,并加速了神经功能的恢复。体外实验证实了Tp53 可通过促进 Klhl4 的转录来增加 Slc7a11 的泛素化水平,在此过程中Kelch结构域对Klhl4识别底物蛋白Slc7a11至关重要。综上所述,研究阐明了Tp53介导的脊髓损伤中神经元铁死亡的机制,为临床转化提供了潜在靶点和启示。
https://orcid.org/0000-0002-5605-9103 (Peigen Xie);
https://orcid.org/0009-0003-6241-4217 (Yong Xuan);
https://orcid.org/0000-0002-4862-5443 (Zongsheng Yin)

 #br#
#br#