中国神经再生研究(英文版) ›› 2026, Vol. 21 ›› Issue (8): 3815-3823.doi: 10.4103/NRR.NRR-D-25-00420
宏基因组组装基因组分析帕金森病患者的肠道菌群特征
Gut microbial community of patients with Parkinson’s disease analyzed using metagenome-assembled genomes
Yi Zhang1, #, Chengjun Mo1, #, Xiaoqin He2, Qin Xiao1, *, Xiaodong Yang1, *
- 1Department of Neurology and Institute of Neurology, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China;
2Department of General Practice, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
摘要:
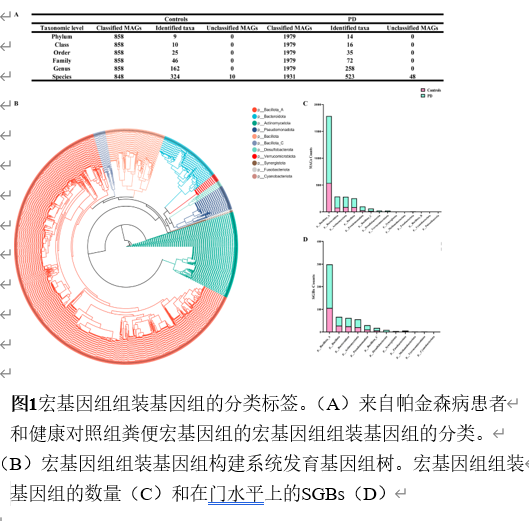
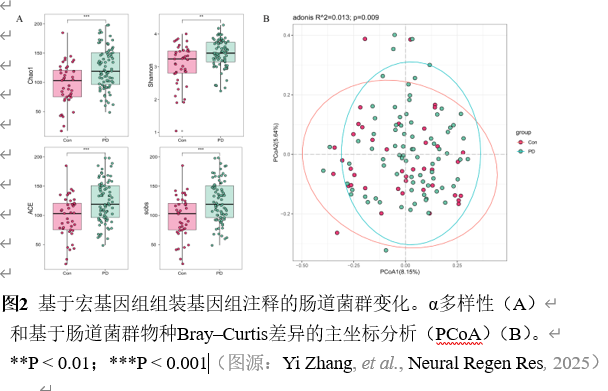
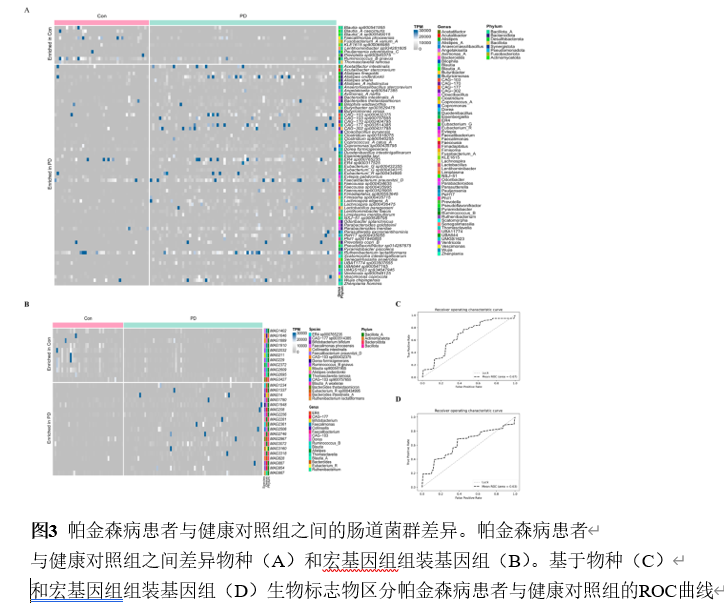
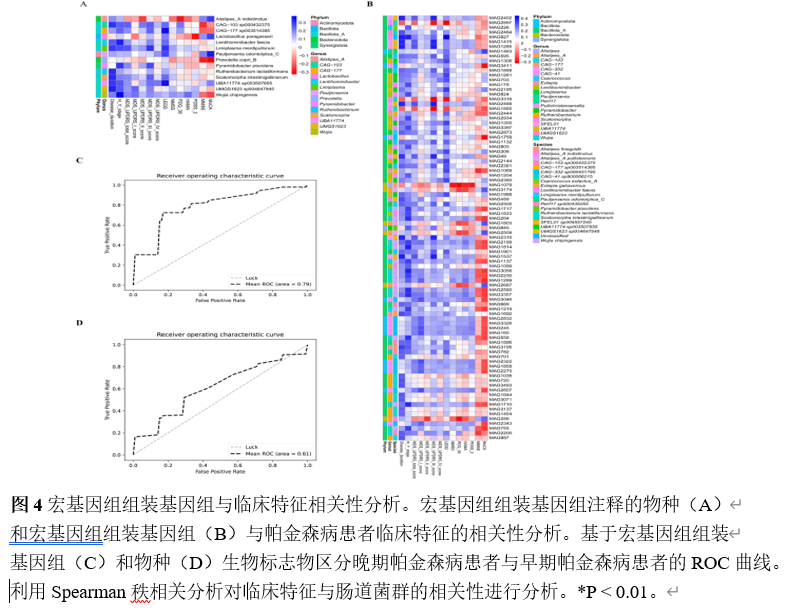
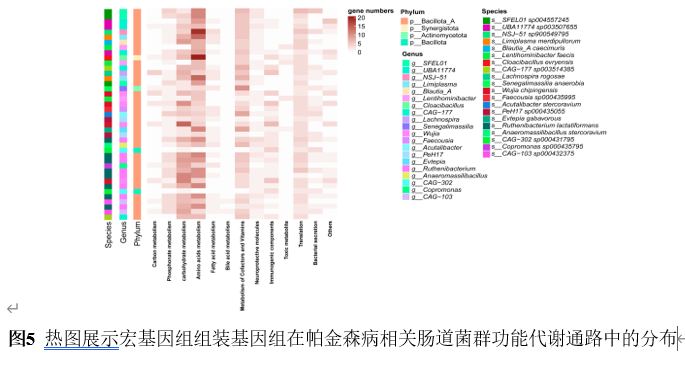
宏基因组分箱为重建稀有群落成员的基因组提供了一种有效的方法。宏基因组组装基因组,即从宏基因组读段组装而成的菌群基因组草图,已被广泛用于研究各种人类疾病中的菌群特征。然而,将肠道菌群宏基因组组装基因组应用于帕金森病患者的研究较为罕见。实验从81例帕金森病患者和41名健康对照的粪便中重建2837个高质量宏基因组组装基因组,在基因组水平探索肠道菌群组成和功能。从宏基因组序列中重建微生物基因组显著提升了微生物基因组的多样性和数量,尤其是不可培养菌株的基因组。分析显示帕金森病患者α多样性显著升高,菌群结构明显分离;578个物种级基因组箱中,354个仅在帕金森病患者检出。随机森林模型证实,30个差异物种与50个差异宏基因组组装基因组均能区分帕金森病与健康人,曲线下面积分别达0.67与0.63。进一步将宏基因组组装基因组与临床特征关联,发现132个与疾病严重程度紧密相关的宏基因组组装基因组,据此构建的模型曲线下面积升至0.79。功能注释揭示这些关键宏基因组组装基因组富集于碳代谢、胆汁酸代谢、神经保护分子及免疫原性通路。通过宏基因组组装基因组与帕金森病临床特征联合分析,识别出帕金森病患者中的肠道微生物群失调。发现132个与疾病严重程度紧密相关的宏基因组组装基因组,据此构建的模型曲线下面积升至0.79。功能注释揭示这些关键宏基因组组装基因组富集于碳代谢、胆汁酸代谢、神经保护分子及免疫原性通路。此研究为在基因组水平上探讨肠道微生物群与帕金森病的关系提供了全面资源,有望深化对该疾病潜在机制的理解。
https://orcid.org/0000-0002-0385-4525 (Xiaodong Yang); https://orcid.org/0000-0001-5554-1443 (Qin Xiao)