RR:中国上海市第一人民医院孙隽然团队提出SLC7A11是视网膜退行性疾病的潜在治疗靶点
年龄相关性黄斑变性(AMD)是一种多因素疾病,是工业化国家不可逆转性视力丧失的主要原因。进行性光感受器细胞死亡是AMD的主要病理特征之一,这一过程将随着疾病的进展导致视力丧失[1]。先前的研究表明铁死亡可能与视网膜退行性疾病有关[2-4]。然而,铁死亡与感光细胞死亡的关系及其具体的分子机制仍不确定。SLC7A11作为胱氨酸/谷氨酸转运蛋白系统的组成部分,其抑制会导致脂质过氧化水平升高和细胞抗氧化能力降低,最终导致铁死亡[5]。然而,光感受器细胞死亡背后的分子过程,铁死亡与感光细胞死亡的关系及其具体的分子机制,SLC7A11对视网膜退行性疾病发生发展的影响,尚不明确。因此,确定其他机制对于开发与光感受器丧失相关的视网膜疾病的新治疗策略至关重要。
中国上海市第一人民医院孙隽然团队在《中国神经再生研究(英文)》(Neural Regeneration Research)2026年第1期发表了研究论文。该研究发现SLC7A11调节的铁死亡是光感受器变性的主要病理过程,通过Nrf2-SLC7A11-GPX4信号通路使得胱氨酸耗竭、铁离子积累和脂质过氧化增强,最终导致感光细胞死亡和随后的视觉功能损伤。该文带来的启示:SLC7A11过表达抑制光诱导视网膜变性(LIRD)小鼠的视网膜变性,这有望提供特异性的抗氧化基因治疗靶点来减轻脂质过氧化引起的感光细胞的损伤。黄晓旭,张宇萌和姜宇欣为论文共同第一作者,孙隽然和赵晓换为论文通讯作者。
光损伤增加光感受器细胞死亡
为了评估光感受器细胞的死亡,在光诱导的小鼠中对视网膜组织进行切片,并进行PI和TUNEL染色。与对照组相比,光诱导视网膜变性(LIRD)组视网膜中PI阳性和TUNEL阳性细胞的数量以时间依赖的方式增加。过量的Fe2+具有高度的细胞毒性,促进铁死亡发生[6]。光照3天后,视网膜中的Fe2+ 浓度和比例显著增加,表明光诱导的光感受器细胞死亡中的铁死亡增加。在建立LIRD实验小鼠模型后,进行ERG实验以进一步评估LIRD后视网膜功能的变化。结果显示,LIRD后3天,a波和b波振幅显著降低,这表明视网膜功能失调。综合以上结果,视网膜结构的病理变化与ERG提示的功能变化相匹配,在LIRD模型中表现出明显的视网膜损伤(图1)。
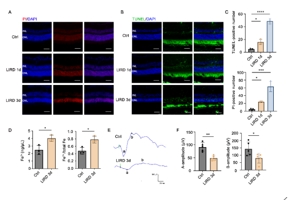
图1 LIRD诱导感光细胞死亡
(图源:Huang et al., Neural Regen Res, 2026)
LIRD增加小鼠模型中视网膜SLC7A11水平
基于GEO数据库数据集的差异表达分析显示,与对照组相比,LIRD组的白化小鼠分别表现出232个基因和45个基因的上调和下调。光损伤后SLC7A11表达模式的热图显示,在LIRD组中,SLC7A11和铁代谢相关蛋白如Homx1、Lcn2和Trf上调。基于数据库结论,为了验证本研究中SLC7A11的表达水平,收集在不同时间点LIRD模型的小鼠视网膜,并进行了蛋白质印迹分析。与对照组相比,LIRD后第1天SLC7A11水平显著升高,第3天降低。同时,RT-PCR分析显示,SLC7A11的mRNA水平在第1天上调,然后下降。免疫荧光结果也显示,LIRD后第1天,SLC7A11在视网膜ONL中的表达显著增加(图2)。
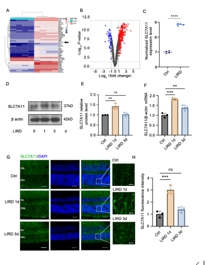
图2 LIRD小鼠视网膜中SLC7A11表达升高
(图源:Huang et al., Neural Regen Res, 2026)
光损伤诱导体内铁死亡和氧化应激
ROS的积累超过了过氧化物酶系统的清除能力会导致氧化应激,引发脂质过氧化,最终导致铁死亡。因此,使用DHE染色测量有或无光暴露的小鼠视网膜中的ROS水平。结果显示光照后第1天和第3天,ROS水平增加。此外,参与氧化应激的几种分子,包括Nrf2和HO-1的表达水平增加,在第3天达到峰值,这表明氧化应激参与了光诱导的光感受器损伤。作为铁死亡的负调节因子,GPX4在降低脂质过氧化物水平方面发挥作用,并且在光照后也观察到GPX4以时间依赖的方式下调。腹腔内注射NaIO3被广泛认为是氧化应激损伤的一种模型[7],在该模型中研究了氧化应激与铁死亡的潜在关联。NaIO3处理后6小时,Nrf2和SLC7A11的表达增加,在第1天趋于平稳,然后下降,而HO-1的表达在第3天趋于平稳然后下降。治疗后第6天至第7天,GPX4的蛋白表达持续下降。为了进一步验证SLC7A11在光感受器变性中的重要性,在小鼠光暴露前连续3天腹腔内注射SLC7A11的抑制剂SAS。SAS处理显著增加了光感受器中TUNEL阳性细胞的数量,并在蛋白质水平下调Nrf1、GPX4和HO-1的表达(图3)。
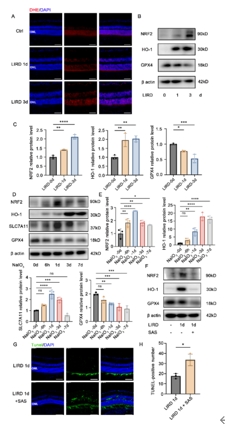
图3 在体内光损伤诱导铁死亡和氧化应激
(图源:Huang et al., Neural Regen Res, 2026)
H2O2处理导致SLC7A11表达上调,SLC7A11抑制剂SAS促进661W细胞铁死亡
为了进一步阐明SLC7A11在光感受器中的机制,首先用不同浓度和时间的H2O2处理661W细胞,并通过蛋白质印迹分析SLC7A11的表达,观察到在H2O2处理后661W细胞中SLC7A11的表达显著上调。BODIPY-C11 581/591检测也显示H2O2处理显著增加了脂质过氧化,具体表现为氧化/非氧化状态的免疫荧光强度比值增加。同样,SAS处理661W细胞,以探索SLC7A11在光感受器变性中的潜在作用。先前的研究表明,SLC7A11通过将半胱氨酸转运到胞质溶胶中来促进GSH的产生,从而减少细胞中的脂质过氧化,从而抑制铁死亡[8]。使用200μM SAS治疗后GSH水平下降,用400μM SAS处理后几乎检测不到GSH水平。CCK-8测定显示,0-400μM SAS单独处理对661W细胞的生存能力没有变化,而SAS与H2O2联合处理导致细胞生存能力呈剂量依赖性降低。在H2O2处理后,GPX4下调,而Nrf2和HO-1上调。使用SAS处理显著抑制了GPX4、Nrf2和HO-1蛋白的表达水平,表明SLC7A11的抑制增强了661W细胞的脂质过氧化,促进了铁死亡(图4)。
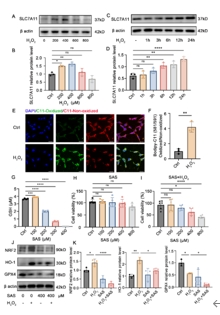
图4 在H2O2处理的661W细胞中,SLC7A11表达上调,并且SLC7A11抑制剂SAS促进661W铁死亡
(图源:Huang et al., Neural Regen Res, 2026)
SLC7A11敲低促进铁死亡,SLC7A11过表达抑制铁死亡
使用SLC7A11的siRNA和过表达质粒来研究SLC7A11在光感受器铁死亡中的作用。首先,使用具有不同序列的三种靶向Slc7a11 siRNA(si-Slc7a11#1、#2或#3)在661W细胞中敲低SLC7A11,siSlc7a11#1和#3都有效地敲低了SLC7A11蛋白表达。RT-PCR分析显示,siSlc7a11#3可能对SLC7A11具有最大的敲除效率。因此,选择siSlc7a11#3进行后续实验。结果,在siSlc7a11转染661W细胞后,细胞活力没有显著变化。因此,Slc7a11 siRNA显著降低了在H2O2暴露后的GPX4、Nrf2和HO-1的蛋白表达。
Fer-1是铁死亡的有效抑制剂[9]。单独的Fer-1并不显著影响661W细胞的生存能力;因此,选择40μM进行后续实验。CCK-8测定显示,在SLC7A11敲低和H2O2暴露后,Fer-1挽救了降低的细胞活力,提供了进一步的证据表明SLC7A11敲低促进661W细胞的铁死亡(图5)。
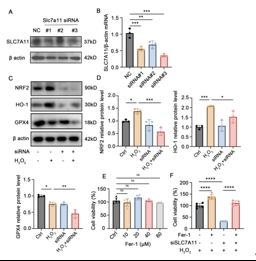
图5 SLC7A11敲低促进661W细胞铁死亡
(图源:Huang et al., Neural Regen Res, 2026)
接下来,将SLC7A11过表达质粒转染到661W细胞中以上调SLC7A11的表达。结果表明,将SLC7A11过表达质粒转染661W细胞后,细胞活力没有显著变化。蛋白质印迹显示,该处理增加了SLC7A11、GPX4、Nrf2和HO-1的表达,并抑制了H2O2诱导的GPX4表达下调,同时抗氧化蛋白(Nrf2、HO-1)水平增强。铁死亡诱导剂Erastin与GPX4抑制剂RSL3一起诱导铁死亡[10]。CCK-8测定显示,在erastin或RSL3诱导的铁死亡过程中,细胞活力呈剂量依赖性降低。SLC7A11过表达减轻了H2O2暴露后erastin或RSL3引起的细胞活力的下降。上述结果表明,SLC7A11过表达有助于在体外抑制光感受器铁死亡(图6)。
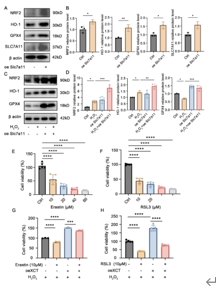
图6 SLC7A11过表达抑制661W细胞铁死亡
(图源:Huang et al., Neural Regen Res, 2026)
SLC7A11过表达抑制光照后视网膜变性
为了验证SLC7A11过表达的治疗潜力,将重组AAV介导的SLC7A11(rAAV2/8-SLC7A11-GFP)转染到小鼠视网膜中,并于两周后接受光照。冰冻切片显示rAAV2/8-SLC7A11-GFP有效转染到视网膜色素上皮(RPE)和整个视网膜范围的光感受器中。通过蛋白质印迹实验证实了外源性SLC7A11和其他相关蛋白在视网膜中的表达。LIRD前和LIRD后3天的ERG测定显示,SLC7A11在光感受器中的过表达挽救了LIRD后三天的a波和b波振幅。通过TUNEL和PI染色研究rAAV2/8-SLC7A11对光感受器细胞死亡的影响,结果显示,在LIRD后3天,SLC7A11的过表达显著降低了TUNEL阳性细胞和PI阳性细胞的数量(图7)。
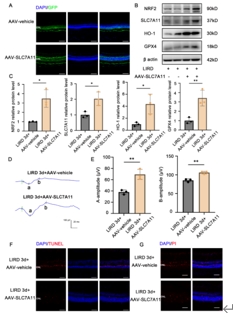
图7 SLC7A11在光感受器中的过表达抑制LIRD小鼠的视网膜变性
(图源:Huang et al., Neural Regen Res, 2026)
综上所述,铁死亡和SLC7A11在光感受器变性中的发挥着关键作用。本研究使用光照和碘酸钠(NaIO3)诱导的视网膜损伤的小鼠模型,研究了SLC7A11对铁死亡的影响。利用小鼠光感受器衍生的细胞系(661W)来阐明固有的分子机制。进一步研究了SLC7A11介导的氧化应激诱导的铁死亡在视网膜变性的进展中发挥作用。因此,SLC7A11可能是视网膜退行性疾病患者的潜在治疗靶点。
原文链接:https://doi.org/10.4103/NRR.NRR-D-23-01741
参考文献
[1] Deng Y, Qiao L, Du M, et al. Age-related macular degeneration:
Epidemiology, genetics, pathophysiology, diagnosis, and targeted therapy. Genes
Dis. 2021;9:62-79.
[2] Tang Z, Ju Y, Dai X, et al. HO-1-mediated ferroptosis as a target for protection
against retinal pigment epithelium degeneration. Redox Biol. 2021;43:101971.
[3] Sun Y, Zheng Y, Wang C, et al.
Glutathione depletion induces ferroptosis, autophagy, and premature cell
senescence in retinal pigment epithelial cells. Cell Death Dis. 2018;9:753.
[4] Shu W, Baumann BH, Song Y, et al. Ferrous but not ferric iron
sulfate kills photoreceptors and induces photoreceptor-dependent RPE
autofluorescence. Redox Biol. 2020;34:101469.
[5] Dong H, Qiang Z, Chai D, et al. Nrf2 inhibits ferroptosis and
protects against acute lung injury due to intestinal ischemia reperfusion via
regulating SLC7A11 and HO-1. Aging (Albany NY). 2020;12:12943-12959
[6] Zhao X, Gao M, Liang J, et al. SLC7A11 reduces laser-induced
choroidal neovascularization by inhibiting RPE ferroptosis and VEGF production.
Front Cell Dev Biol. 2021;9:639851.
[7] Chan CM, Huang DY, Sekar P, et al.
Reactive oxygen species-dependent mitochondrial dynamics and autophagy confer
protective effects in retinal pigment epithelial cells against sodium
iodate-induced cell death. J Biomed Sci. 2019;26:40.
[8] Fang X, Cai Z, Wang H, et al. Loss of cardiac ferritin H facilitates cardiomyopathy via
Slc7a11-mediated ferroptosis. Circ Res. 2020;127:486-501.
[9] Miotto G, Rossetto M, Di Paolo ML, et al. Insight into the mechanism of ferroptosis inhibition by
ferrostatin-1. Redox Biol. 2020;28:101328.
[10] Chen X, Kang R, Kroemer G, et al. Broadening horizons: the role of ferroptosis in cancer. Nat
Rev Clin Oncol. 2021;18:280-296.