NRR:“小基因大作为”:海军军医大学长征医院周许辉团队揭示RNA修饰相关标志物可作为脊髓损伤精准诊疗新靶点#br#
#br#
作者:黄世学(第一作者)、焦坤(共同第一作者)、李可清(共同第一作者)周许辉(通讯作者)周鑫(共同通讯作者)
#br#
脊髓损伤(Spinal Cord Injury, SCI)是现代社会中一种常见的中枢神经系统损,其给家庭、经济和社会带来了巨大的负担。据估计,全球SCI的年发病率在25万至50万例,其中中国每年约有66,374例新发病例,北美每年约有17,000例新发病例[1-2]。临床治疗脊髓损伤常用的方法有手术减压、激素冲击疗、高压氧治疗等[3]。然而,脊髓损伤的复杂性阻碍了治疗方法的发展,靶向诊疗生物标志物的筛选和鉴定迫在眉睫[4]。表观遗传学是指在不改变核苷酸序列的情况下基因表达的可遗传变化,包括DNA甲基化、组蛋白修饰和RNA修饰。在SCI中,mRNA修饰状态的变化与神经细胞存活、轴突再生、神经修复和炎症反应等重要过程密切相关[5-6]。然而,脊髓损伤发生前后发生显著修饰变化的关键RNA仍然未知。此外,差异RNA在SCI后发挥作用的细胞微环境也有待探索。#br#
#br#
海军军医大学长征医院周许辉/周鑫团队在《中国神经再生研究(英文版)》(Neural Regeneration Research)发表的研究中,通过整合公共bulk-RNA 测序与单细胞转录组数据,发现脂质-糖代谢交叉调控的四个RNA修饰相关基因(Elovl6、Idi1、Sqle、Stbd1)在脊髓损伤(SCI)后呈显著差异表达,可作为高准确性诊断标志物(ROC-AUC>0.7);其中Elovl6和Sqle与巨噬/小胶质细胞极化密切相关,Stbd1则与神经元糖噬活性增强有关。进一步的机器学习建模、诺莫图构建及分子对接实验证实,老药乙酰氨基酚可与四基因靶蛋白稳定结合,提示其具有重定位为SCI急性期治疗药物的潜力。该研究带来的启示:RNA修饰-代谢重编程轴可能是调控SCI免疫微环境及神经元命运的新靶点,为开发可穿透血脑屏障的生物标志物及小分子干预策略提供了理论与实验依据。黄世学、焦坤、李可清为共同第一作者,周鑫、周许辉教授为通讯作者。#br#
#br#
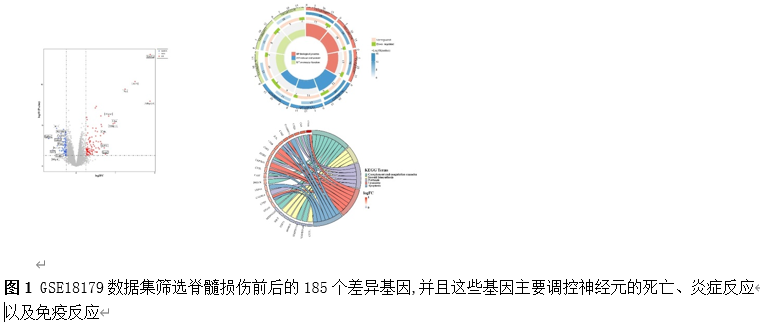
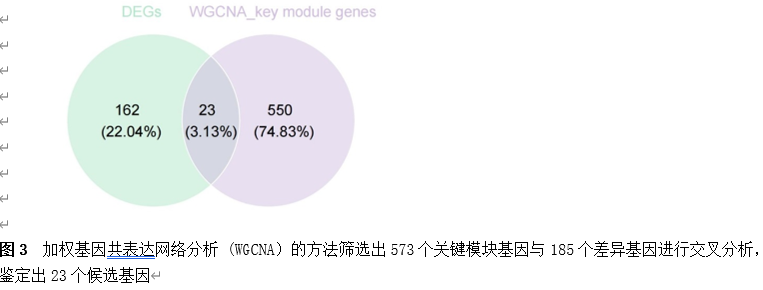
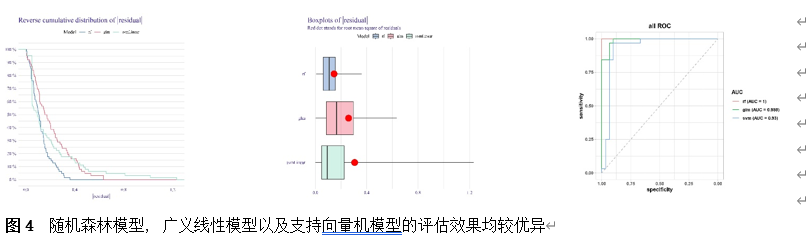
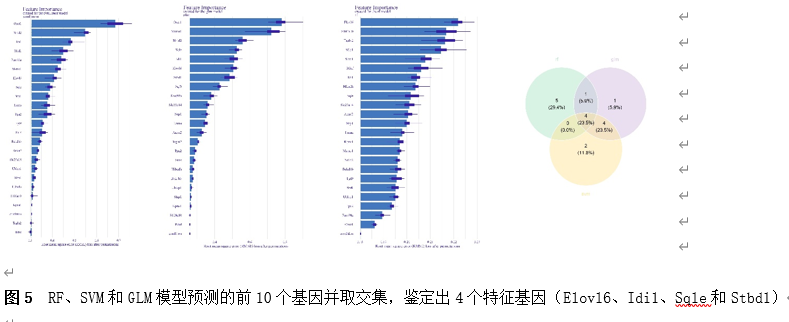
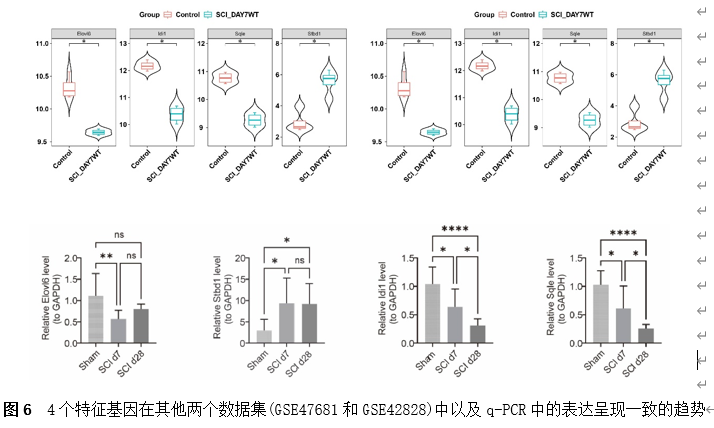
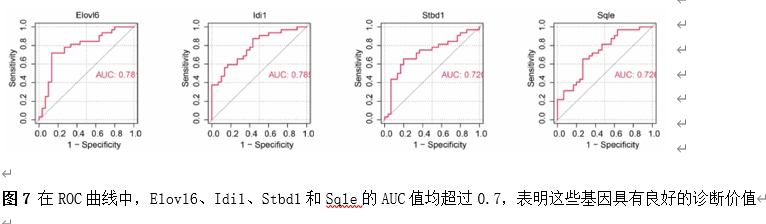
筛选差异基因是脊髓损伤靶向诊疗的关键。因此,该研究首先使用Gene Expression Omnibus(GEO)数据库中的GSE18179数据集筛选脊髓损伤前后的差异基因,并对其进行Gene Ontology(GO)和Kyoto Encyclopedia of Genes and Genomes(KEGG)分析。总共筛选出185个差异基因,并且这些基因主要调控神经元的死亡、炎症反应以及免疫反应等(图1)。此外,单样本基因集富集分析 (ssGSEA)提示,SCI后RNA修饰相关基因(RRGs)评分明显较低(图2)。随后,作者应用加权基因共表达网络分析 (WGCNA)的方法筛选出573个关键模块基因。与185个差异基因进行交叉分析,鉴定出23个候选基因(图3)。为了更高效准确地筛选出SCI后RNA修饰相关的基因,运用了三种机器学习模型(随机森林模型, 广义线性模型以及支持向量机模型)。这些模型均能表现出类似的优异性能(图4)。根据均方根误差分配给候选基因的重要性得分,选择RF、SVM和GLM模型预测的前10个基因。随后,通过上述3个模型中前10个基因的交集,鉴定出4个特征基因(Elovl6、Idi1、Sqle和Stbd1)(图5) 。在GSE18179中,Elovl6、Idi1和Sqle的表达量显著降低,而Stbd1在SCI组内的表达量显著增加。这些基因在SCI前后的表达模式在其他两个数据集(GSE47681和GSE42828)中以及q-PCR中也得到了验证(图6) 。与此同时, 在ROC曲线中,Elovl6、Idi1、Stbd1和Sqle的AUC值均超过0.7,表明这些基因具有良好的诊断价值,可能作为生物标志物(图7)。

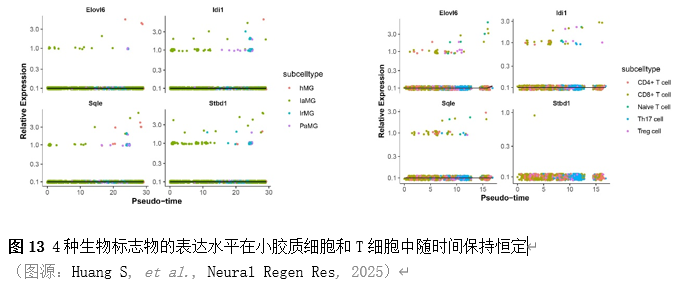
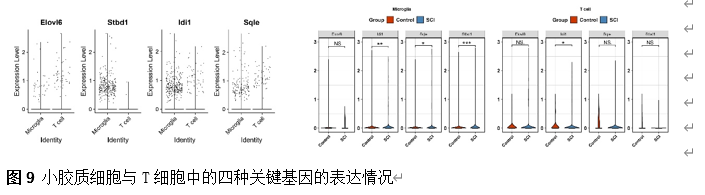
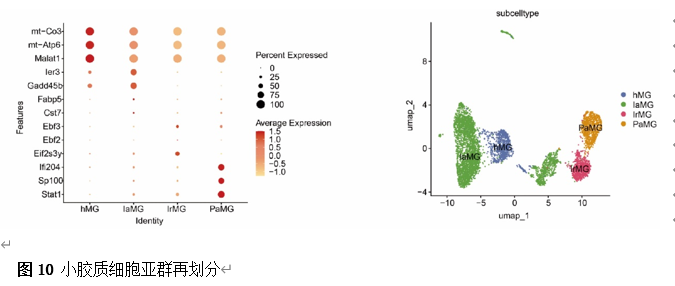
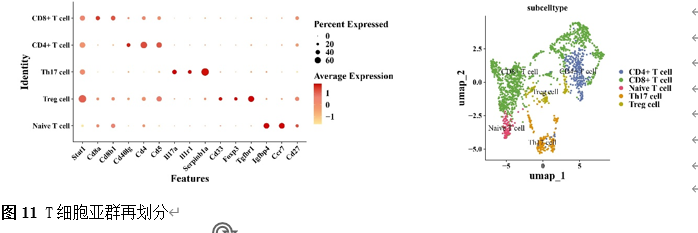
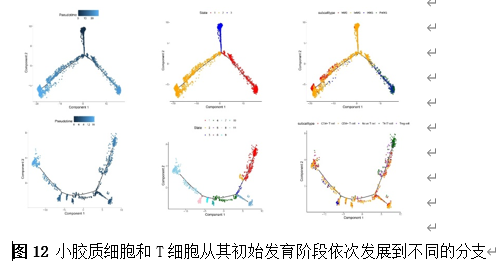
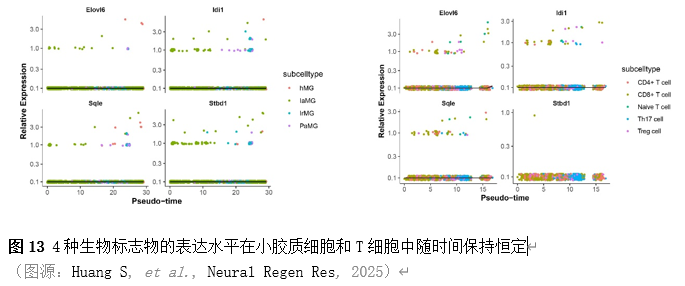
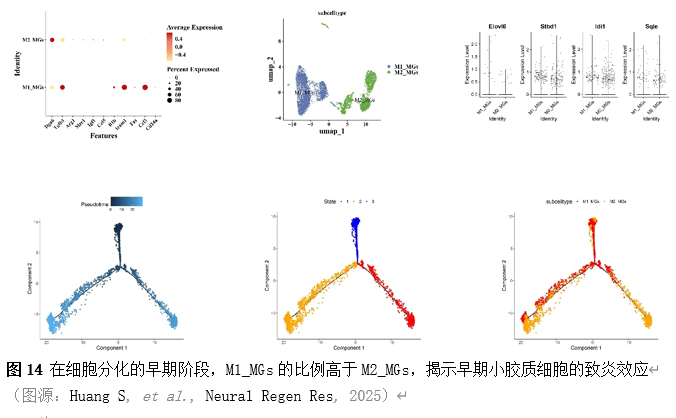
确定差异基因表达的细胞和微环境能够有效推进SCI的诊治研究。因此,该研究团队对公共数据库单细胞测序数据集GSE182803进行一系列分析。通过使用气泡图和热图,可视化每种细胞类型的重要标记基因的表达(图8)。值得注意的是,与对照组相比,SCI组表现出更高的中性粒细胞、单核细胞、成纤维细胞、T细胞和DC百分比,而小胶质细胞和B细胞的百分比较低(图8)。其中,SCI组表现出小胶质细胞的存在显着减少和T细胞的存在显着增加,从而将这两种细胞类型确立为差异细胞(图8)。进一步分析表明,Elovl6在小胶质细胞和T细胞中均均匀表达,而Idi1、Sqle和Stbd1主要在小胶质细胞中表达(图9)。SCI组和对照组小胶质细胞中Idi1、Sqle和Stbd1的表达存在差异。此外,Idi1 在这些组的 T 细胞中表现出差异表达。因此,小胶质细胞和T细胞被确定为关键细胞(图9)。作者进一步将小胶质细胞分为6个亚群,并指定为4种不同的细胞亚型:稳态小胶质细胞(hMG)、干扰素反应性小胶质细胞(IrMG)、损伤相关小胶质细胞(IaMG)和增殖相关小胶质细胞(PaMG)(图10)。同样,T细胞被分为7个亚群,并分为5种细胞亚型:CD4+T细胞、CD8+T细胞、幼稚T细胞、Th17细胞和调节性T(Treg)细胞(图11)。此外,小胶质细胞和T细胞从其初始发育阶段依次发展到不同的分支 (图12)。此外,发现4种生物标志物(Elovl6、Idi1、Sqle和Stbd1)的表达水平在小胶质细胞和T细胞中随时间保持恒定(图13)。为了了解数据集中每个细胞在细胞状态转换过程中所经历的基因表达变化,对小胶质细胞进行了降维和聚类分析。他们被重新聚集到不同的亚组中。结果表明,小胶质细胞分为6个亚组,并注释为2个细胞亚型:M1_MGs和M2_MGs(图14)。接下来,为了了解scRNA-seq数据集中每个细胞在细胞状态转换过程中所经历的关键基因表达变化,对这些关键细胞簇进行了细胞假时间轨迹分析。结果表明,小胶质细胞从发育起始位置依次发育为不同的分支,具有3个不同的发育阶段。在同一发育阶段存在多个细胞簇(图14)。在细胞分化的早期阶段,M1_MGs的比例高于M2_MGs,揭示早期小胶质细胞的致炎效应(图14)。

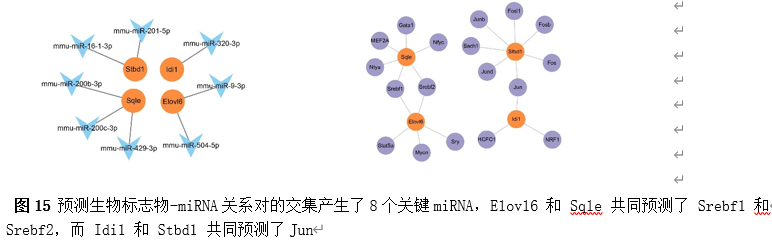
为了进一步了解生物标志物的调控因素,构建了分子调控网络。来自miRanda和miRDB数据库的预测生物标志物-miRNA关系对的交集产生了8个关键miRNA,例如mmu-miR-201-5p(图15)。此外,发现Elovl6、Idi1、Sqle和Stbd1分别预测5、3、6和7个TFs(图15)。值得注意的是,Elovl6 和 Sqle 共同预测了 Srebf1 和 Srebf2,而 Idi1 和 Stbd1 共同预测了Jun。
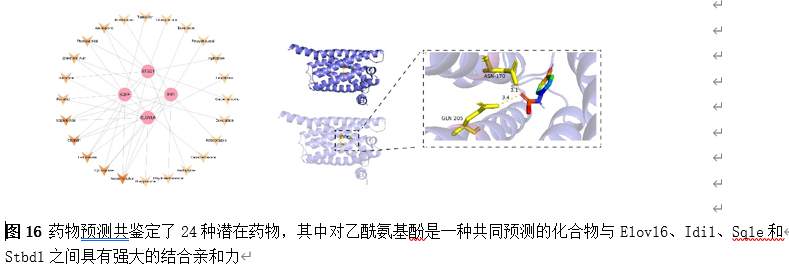
为了筛选可能调控Elovl6、Idi1、Sqle和Stbd1的小分子药物,以支持这些基因在SCI中的表观遗传调控潜力,我们通过药物预测共鉴定了24种潜在药物,其中对乙酰氨基酚是一种共同预测的化合物(图16)。因此,为了确定预测药物与关键基因之间的结合能力,对预测药物和关键基因进行了分子对接。对乙酰氨基酚被选为重点药物。对乙酰氨基酚与Elovl6、Idi1、Sqle和Stbd1结合的自由能分别为-6.3 kcal/mol、-6.1 kcal/mol、-6.1 kcal/mol和-5.4 kcal/mol(≤-5 kcal/mol 的结合能阈值表明具有强大的结合亲和力)。结果表明,对乙酰氨基酚与Elovl6、Idi1、Sqle和Stbd1之间具有强大的结合亲和力。其中,Elovl6和对乙酰氨基酚表现出最高的结合自由能,因此值得选择进行论证。
总之,该研究使用生物信息学分析鉴定了与SCI相关的RNA修饰相关的生物标志物(Elovl6、Idi1、Sqle和Stbd1)。此外,该研究确定了脊髓损伤中的两个关键细胞,T细胞和小胶质细胞,并分析了这些生物标志物在免疫浸润肥大细胞、T细胞和小胶质细胞中的表达模式。功能富集分析揭示了与Elovl6、Idi1、Sqle和Stbd1相关的通路,强调了它们在脊髓损伤病理生理中的潜在作用。这些发现表明,这些生物标志物有望在脊髓损伤的治疗应用中发挥作用。然而,目前的研究还存在一些局限性。首先,在研究中使用异氟醚麻醉可能会对细胞转录组产生干扰。未来的研究可以通过比较不同的麻醉方法,建立麻醉对照组,或采用生物信息学方法来纠正数据,从而排除麻醉效果的影响。此外,该研究还缺乏对人体样本的验证。未来的工作应该纳入人类SCI数据集(如来自GEO或TCGA的数据)和临床样本(如血液或脑脊液),以验证Elovl6、Idi1、Sqle和Stbd1等生物标志物的诊断和治疗潜力。最后,该研究没有在蛋白水平上验证关键分子靶点的表达和功能。未来的研究应采用功能实验,如免疫荧光染色、基因敲除或过表达研究,进一步阐明这些分子在脊髓损伤中的具体机制。这些改进将提高研究结论的普遍性和临床转化价值。
原文链接:https://doi.org/10.4103/NRR.NRR-D-25-00080
参考文献#br#
#br#
[1] ADDIN ZOTERO_BIBL {"uncited":[],"omitted":[],"custom":[]} CSL_BIBLIOGRAPHY [1] Khorasanizadeh M, Yousefifard M, Eskian M, et al. Neurological recovery following traumatic spinal cord injury: a systematic review and meta-analysis. J Neurosurg Spine. 2019;30(5):683-699.#br#
[2] Hu X, Xu W, Ren Y, et al. Spinal cord injury: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther. 2023;8(1):245.#br#
[3] Liu X, Zhang Y, Wang Y, et al. Inflammatory Response to Spinal Cord Injury and Its Treatment. World Neurosurg. 2021;155:19-31.#br#
[4]. Huang S, Zhang Y, Shu H, et al.Advances of the MAPK pathway in the treatment of spinal cord injury. CNS Neurosci Ther. 2024;30(6):e14807.#br#
[5] Hong JY, Davaa G, Yoo H, et al.Ascorbic Acid Promotes Functional Restoration after Spinal Cord Injury Partly by Epigenetic Modulation. Cells. 2020;9(5):1310.#br#
[6] Xie J, Herr S, Ma D, et al.Acute Transcriptomic and Epigenetic Alterations at T12 After Rat T10 Spinal Cord Contusive Injury. Mol Neurobiol. 2023;60(5):2937-2953.#br#