NRR:福建医科大学附属第一医院许卫红团队报道脊髓损伤过程中不同调节性细胞死亡的机制
脊髓损伤是一种受到全球关注的灾难性疾病。脊髓损伤不仅造成运动和感觉的丧失,还导致多种全身并发症[1]。脊髓损伤具有高致残率和高死亡率,目前仍无法治愈。患者一生都需要大量的专业的医疗护理,这给患者、家庭和社会带来巨大的经济和精神负担。目前脊髓损伤的管理策略包括手术减压、药物支持和康复治疗,然而效果并不显著。
脊髓损伤在病理上可分为原发性损伤和继发性损伤[2]。原发性损伤中的脊髓细胞多出现意外细胞死亡, 造成无法逆转的细胞损害和组织坏死,而继发性损伤中脊髓细胞多出现调节性细胞死亡并具有逆转的可能。因此脊髓损伤的治疗重点之一在于如何早期、快速、有效着调控调节性细胞死亡继而减少继发性损伤[3]。近年来新发现的调节性细胞死亡类型如焦亡、铁死亡以及不断更新的自噬在脊髓损伤中的研究受到广泛关注[4-6]。总结脊髓损伤中焦亡、铁死亡和自噬的机制、共同调控途径以及相互作用等方面的研究进展,可能为脊髓损伤的治疗提供新思路。
来自中国福建医科大学附属第一医院许卫红团队在《中国神经再生研究(英文)》(Neural Regeneration Research)上发表了题为“Pyroptosis, ferroptosis, and autophagy in spinal cord injury: regulatory mechanisms and therapeutic targets”的综述,总结了脊髓损伤中焦亡、铁死亡和自噬的作用机制、调控通路和治疗靶点等多方面的研究进展。文章认为多种调节性细胞死亡结合多种调控途径的整体网络将是一种治疗脊髓损伤的潜在多靶点联合方案。
许卫红等总结了焦亡、铁死亡和自噬以及多种调控焦亡、铁死亡和自噬的信号通路参与脊髓损伤的致病机制(图1)。
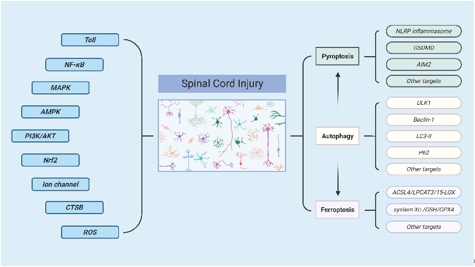
图1脊髓损伤中调节性细胞死亡的信号通路及其调控通路(图源:Zheng et al., Neural Regen Res, 2025)
细胞焦亡可分为激活caspase-1的典型信号通路和激活caspase-4/-5/-11的非典型信号通路[7]。NLRP3炎症小体的成熟包括启动和激活步骤。初级信号可通过TLR结合DAMP/PAMP随后激活NF-κB信号通路来诱导NLRP3炎症小体的启动。次级信号包括K+和Ca2+通道的打开、溶酶体释放组织蛋白酶B(CTSB)、线粒体DNA以及活性氧的产生。初级和次级信号共同启动NLRP3炎症小体的活化,翻译后修饰进一步促进炎症小体复合物的组装。NLRP3/ASC/pro-caspase-1/caspase-1轴是NLRP3炎症小体的激活轴[8]。成熟的caspase-1有2种主要作用途径。其一,caspase-1的p20/p10亚基切割前白细胞介素1β和前白细胞介素18,释放白细胞介素1β和白细胞介素18并引发炎症反应。其二,caspase-1裂解并激活GSDMD,从而引发细胞膜上的寡聚化反应以形成孔隙最终导致细胞死亡[9](图2)。
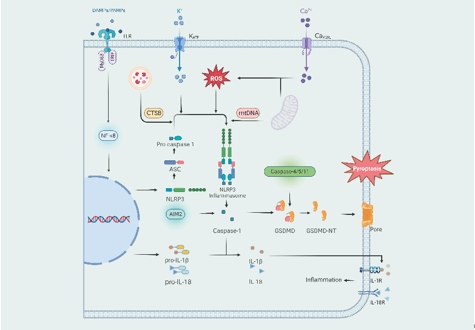
图2焦亡信号通路(图源:Zheng et al., Neural Regen Res, 2025)
在脊髓损伤中,多种类型的脊髓细胞(如神经元、小胶质细胞、星形胶质细胞)已被报道发生焦亡机制。焦亡的关键蛋白或蛋白复合物包括NLRP炎症小体、GSDMD和AIM2,用化合物干扰这些靶点可改善脊髓损伤。例如,脊髓损伤能够诱发NLRP1,ASC和 caspase-1表达增加并激活炎症小体来加重神经元死亡,而血红素氧化酶(HO-1)能通过抑制ATF4来降低NLRP1的表达起到保护神经元作用。脊髓损伤促使NLRP3炎症小体激活并发挥效应损害神经元,使用BAY 11-7082抑制NLRP3炎症小体活化后发现存活神经元数量明显增加。此外,还有靶向GSDMD,AIM2以及其他靶点。
许卫红等总结了脊髓损伤中调节焦亡的信号通路。(1)Toll样信号通路。TLR4可通过JAK2/STAT1/DDX3X/NLRP3信号通路激活NLRP3炎症小体,引起脊髓损伤小鼠脊髓细胞焦亡。(2)NF-κB通路。NF-κB是NLRP3表达所需的关键信号分子。在BV2小胶质细胞中,脱氧雪腐镰刀菌烯醇(deoxynivalenol)通过激活NF-κB通路促进NLRP3炎症小体和白细胞介素1β的表达。(3)MAPK信号通路。Fanconi贫血C组互补组基因(FANCC)过表达能抑制p38/NLRP3通路进而抑制焦亡。(4)离子通道。① K+:细胞内低钾能激活NLRP3炎症小体并促进白细胞介素1β和白细胞介素18的释放。② Ca2+:钙动员在焦亡机制中起关键作用,毒胡萝卜素通过阻断细胞外Ca2+的进入来减少NLRP3炎症小体的激活。③ Mg2+:Mg2+通过抑制钙通道阻止Ca2+内流从而阻断焦亡。④ Zn2+:Zn2+在脊髓损伤中可通过XIST/miR-374a-5p轴使NLRP3炎症小体失活。⑤ Al3+:Al3+可通过DDX3X-NLRP3炎症小体信号通路参与神经元细胞的焦亡和炎症反应。(5)CTSB:能激活NLRP3炎症小体促进焦亡。(6)活性氧:是NLRP3炎症小体在焦亡过程中的直接激活剂,活性氧与NLRP3炎症小体之间存在正循环。(7)AMPK信号通路:二甲双胍通过调节AMPK/NLRP3通路减少焦亡和促炎因子的释放。(8)Nrf2信号通路:Nrf2/ARE轴不仅能抑制NOX4/活性氧/NF-ĸB轴以减少活性氧产生,也能下调NLRP3、caspase-1水平以抑制焦亡。(9)自噬和焦亡的串扰:雷帕霉素通过增强自噬机制来降低caspase-1、GSDMD,白细胞介素1β和白细胞介素18的表达以抑制焦亡。(10)PI3K/AKT信号通路:麦芽酚可通过抑制 PI3K/AKT/NF-κB 通路来抑制 NLRP3 炎症小体的激活。然而,CD73可通过促进PI3K/AKT/FOXO1通路来减轻焦亡。文章通过总结调控焦亡的信号通路和靶点加深了人们对脊髓损伤的机制和治疗的理解。
铁稳态失衡是铁死亡的核心。转铁蛋白与细胞外的Fe3+结合并通过转铁蛋白受体进入细胞质,与转铁蛋白结合的铁通过核内体(endosome)释放Fe3+。Fe3+通过STEAP3还原成Fe2+,Fe2+通过DMT1进入铁池或通过PCBP储存在铁蛋白(FTL/FTH1)中。NCOA4促使铁蛋白降解并释放Fe2+。携带 Fe2+的PCBP2与SLC40A1结合可将铁排出体外[10]。脂质过氧化是铁死亡的标志。Fe2+和H2O2通过Fenton反应产生 活性氧,而Fe3+通过 Haber-Weiss反应还原为Fe2+。活性氧 通过非酶和酶促反应攻击多不饱和脂肪酸。非酶促途径是-OH、O2与多不饱和脂肪酸反应生成PLOOH,酶促途径是花生四烯酸和花生四烯酸在ACSL4,LPCAT3,Fe2+和LOX的作用下生成PLOOH。PLOOH降解为丙二醛和4-HNE最终导致铁死亡[11]。抗氧化机制是抑制铁死亡的调节系统。system Xc-/GSH/GPX4轴:GPX4可抑制活性氧,并将PLOOH还原成PLOH,从而抑制铁死亡[12]。FSP1/CoQ10/NADPH轴:FSP1通过NADPH催化CoQ10形成CoQH2,并消耗NADPH以抑制活性氧的产生[13]。GCH1/BH4/DHFR轴:GCH1可促进BH4的合成,而BH4可促进CoQ10合成从而抵制铁死亡[14](图3)。
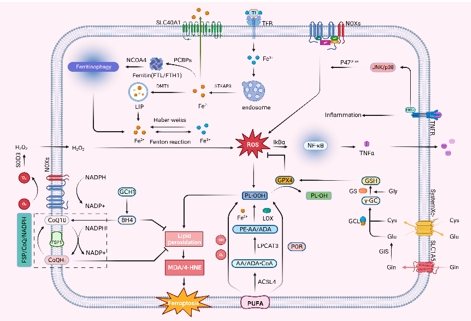
图3铁死亡信号通路(图源:Zheng et al., Neural Regen Res, 2025)
在脊髓损伤中,铁死亡的关键信号通路包括ACSL4/LPCAT3/15-LOX和system Xc-/GSH/GPX4信号轴,调整这些信号轴可以促进受损脊髓的恢复。(1)靶向ACSL4/LPCAT3/15-LOX轴。原花青素能明显降低ACSL4和ALOX15的水平,增加GSH,GPX4,Nrf2和HO-1的表达,从而抑制铁死亡来促进受损脊髓的修复。(2)靶向System Xc-/GSH/GPX4轴。铁死亡抑制剂(SRS16-86)可通过上调system Xc-,GSH和GPX4来减轻铁死亡从而促进脊髓损伤大鼠的恢复。
许卫红等总结了脊髓损伤中调节铁死亡的信号通路。(1)Toll样信号通路。TLR4可降低SLC7A11和GPX4的表达,而TAK-242可抑制TLR4-p38-MAPK通路从而抵抗铁死亡来发挥神经保护作用。(2)NF-κB通路可以通过下调ATF4和SLC7A11的表达来促进铁死亡。(3)离子通道。①Ca2+。铁超载会导致不受控制的钙信号激增,铁/钙失衡会导致神经元损伤和死亡。②Zn2+。锌能通过Nrf2/HO-1通路抑制铁死亡。③Al3+。氧化铝纳米颗粒可通过激活IFN-γ/ASK1/JNK轴加剧铁死亡。(4)CTSB是细胞器特异性启动铁死亡的执行者。铁依赖性溶酶体通透性的增强会释放CTSB导致铁死亡。(5)活性氧在脊髓损伤大鼠体内的积累会导致铁死亡。(6)自噬和铁死亡的串扰。铁死亡过程中的氧化应激、活性氧和代谢产物的生成可诱导自噬,自噬的过度激活也会驱动铁死亡。(7)PI3K/AKT信号通路。LXA4可通过激活PI3K/AKT/Nrf2/HO-1通路抑制铁死亡发挥神经保护作用。(8Nrf2信号通路能通过影响铁储存、运输和代谢来调节铁稳态。(9)MAP信号通路。赖氨酰氧化酶通过激活LysOX/ERK/ALOX5轴诱导神经元铁死亡。而褪黑素可通过与MT2受体结合激活ERK/Nrf2通路从而抑制铁死亡。JNK和P38通路同样调控铁死亡途径。(10)AMPK信号通路。AMPK介导的BECN1磷酸化可通过阻断system Xc-的活性来促进铁死亡。相反地,AMPK激活通过诱导乙酰辅酶A羧化酶磷酸化来抑制铁死亡。
自噬的形成通常需要5个步骤:启动、成核、伸长、自噬体(autophagosome)-溶酶体融合和降解[15]。(1)启动。ULK1可与ATG13,ATG17和ATG101形成ULK1复合物。(2)成核。ULK1复合物激活VPS34和BECN1,并与VPS15结合形成PI3K核心复合物,后者分别与ATG14和UVRAG结合形成PI3KC3-C1和PI3KC3-C2。ULK1复合物可磷酸化ATG9并启动转运,ATG9进一步促进PI3KC3-C1,ATG21,ATG2-ATG18和ATG12-ATG5-ATG16复合物的招募,以形成PI3P。(3)延伸。PI3P上的WIPI1与ATG2结合,促进脂质运输。WIPI2可招募E3酶,促进吞噬体(phagophore)扩张。ATG12通过E1酶ATG7和E2酶ATG10与ATG5和ATG16结合,形成ATG12-ATG5-ATG16复合物。LC3在ATG4和ATG7的作用下形成膜结合脂化的LC3-I,并在E2酶ATG3和E3酶ATG12-ATG5-ATG16复合物的催化下与PE结合形成铆接的LC3-II。(4)自噬体-溶酶体融合。STX17在IRGM和ATG8的帮助下形成ARP,使STX17能够插入自噬体膜。HOPS由VPS11,VPS16,VPS18,VPS33,VPS39和VPS41组成。HOPS通过Vps39和Vps41与自噬体和溶酶体上的Rab7相耦合。自噬体膜上的SNAP29和STX17与溶酶体上的VAMP8和HOPS上的VPS33结合,形成了一个以HOPS为杠杆、SNARE为拉链的膜融合模型。(5)降解。成熟的自噬体与溶酶体融合,形成自噬性溶酶体(autolysosome),导致自噬体内膜和结合的自噬物质降解(图4)。

图4自噬信号通路(图源:Zheng et al., Neural Regen Res, 2025)
自噬参与了脊髓损伤大鼠的轴突变性。ULK1在脊髓损伤后迅速升高,自噬体水平显著增加。VEGF165可上调beclin-1来增强自噬促进脊髓恢复。维甲酸可增加脊髓损伤大鼠LC3-II的表达,有效诱导自噬通量促进神经元修复。二甲双胍可通过促进AMPK和抑制mTOR通路,导致p62的消耗和并促进自噬通量,促进脊髓恢复。自噬产生有利/不利的结果,取决于细胞的类型以及发生自噬的环境和时期。对脊髓损伤中自噬相关靶点的深入探讨能为脊髓损伤治疗提供方向。
许卫红等总结了脊髓损伤中调节自噬的信号通路。(1)AMPK信号通路通过多种途径在启动、成核、伸长和融合整个过程来正向调节自噬。(2)离子通道。①K+。溶酶体K+通道TMEM175的敲除会导致自噬体-溶酶体融合阶段的异常。②Ca2+。溶酶体通过电压门控钙通道释放钙是溶酶体与自噬体融合所必需的。③Mg2+。Mg2+可通过AMPK/mTOR通路促进自噬。然而,高Mg2+也会抑制自噬。④Zn2+。适量的锌可通过积极调节自噬保护脊髓,而锌中毒则会加剧神经元损伤。⑤Al3+。麦芽糖酸铝通过降低LC3-II/I比率和p62水平来抑制自噬,穿心莲内酯可改善麦芽糖酸铝诱导的神经毒性。(3)CTSB抑制剂是治疗多种神经系统疾病的候选药物。(4)活性氧能够对自噬进行全过程调节。(5)PI3KC1/AKT/mTORC1信号通路。mTORC1通路调节自噬的启动、核化、延伸以及与溶酶体的融合。(6)Nrf2通路通过p62与自噬密切相关,p62可竞争性结合Keap1,导致Nrf2积累并激活下游通路。(7)Toll样信号通路对自噬具有正/负调控作用。(8)NF-κB信号通路也可在不同情况下对自噬进行正/负调节。(9)MAPK信号通路。双过氧钒可激活ERK1/2通路,促进 脊髓损伤 大鼠的自噬机制保护神经元。JNK1对Bcl-2的多位点磷酸化可提高beclin-1的水平从而促进自噬。P38抑制剂(SB203580)能促进自噬通量以保护神经元。总之,通过调节自噬通路能提高脊髓细胞的存活率并促进脊髓损伤后的组织修复。
许卫红等总结了脊髓损伤中焦亡、铁死亡和自噬的潜在机制、调控通路、串联途径、治疗靶点和相关药物。多种调节性细胞死亡类型以及多种调节性细胞死亡的调节途径参与了脊髓损伤的病理机制。探索同时调控调节性细胞死亡的多种途径并确立多靶点联合治疗方案是一个非常有趣的研究方向, 未来的研究应完善多种调节性细胞死亡与多种调控通路相结合的整体网络。进一步探讨在脊髓损伤的不同时期是否存在不同调节性细胞死亡类型的激活顺序。进一步研究自噬在脊髓损伤的不同的损伤程度和损伤持续时间的情况下调控其他调节性细胞死亡类型的相关机制将是非常有意义的。当然该研究也存在一定性局限性。焦亡、铁死亡和自噬在脊髓损伤中的作用十分复杂,并且通过调节调节性细胞死亡的调控途径对脊髓损伤的影响尚未完全阐明。此外,有关脊髓损伤中的调节性细胞死亡的研究大多局限于基础实验,未来还需要更多的临床研究来总结经验并进一步完善综述。总之,无论哪种调节性细胞死亡类型,都不可能是单独在脊髓损伤中发挥决定性作用。人们需要多靶点联合来治疗脊髓损伤,还需要针对脊髓损伤 的不同阶段使用相关药物进行个性化的现代医疗管理。这篇综述为探索多种调节性细胞死亡和调控通路在脊髓损伤中的作用提供了见解,并为进一步实现脊髓损伤的多靶点联合治疗带来启发与展望。
原文链接:https://doi.org/10.4103/NRR.NRR-D-24-00112
参考文献
[1] Ding W, Hu S, Wang P, et al. Spinal cord injury: the global incidence, prevalence, and disability From the Global Burden of Disease Study 2019. Spine (Phila Pa 1976). 2022;47(21):1532-1540.
[2] Alcántar-Garibay OV, Incontri-Abraham D, Ibarra A. Spinal cord injury-induced cognitive impairment: a narrative review. Neural Regen Res. 2022;17(12):2649-2654.
[3] Shi Z, Yuan S, Shi L, et al. Programmed cell death in spinal cord injury pathogenesis and therapy. Cell Prolif. 2021;54(3):e12992.
[4] Hu X, Chen H, Xu H, et al. Role of pyroptosis in traumatic brain and spinal cord injuries. Int J Biol Sci. 2020;16(12):2042-2050.
[5] Li QS, Jia YJ. Ferroptosis: a critical player and potential therapeutic target in traumatic brain injury and spinal cord injury. Neural Regen Res. 2023;18(3):506-512.
[6] Ray SK. Modulation of autophagy for neuroprotection and functional recovery in traumatic spinal cord injury. Neural Regen Res. 2020;15(9):1601-1612.
[7] Yang J, Liu Z, Xiao TS. Post-translational regulation of inflammasomes. Cell Mol Immunol. 2017;14(1):65-79.
[8] Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707-735.
[9] Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111-116.
[10] Chen X, Li J, Kang R, et al. Ferroptosis: machinery and regulation. Autophagy. 2021;17(9):2054-2081.
[11] Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482(3):419-425.
[12] Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019;133:144-152.
[13] Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693-698.
[14] Kraft VaN, Bezjian CT, Pfeiffer S, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6(1):41-53.
[15] Melia TJ, Lystad AH, Simonsen A. Autophagosome biogenesis: from membrane growth to closure. J Cell Biol. 2020;219(6):e202002085.