NRR:中国广州医科大学徐评议团队报道了ATP6V0A1在帕金森病中调控自噬保护多巴胺神经元的作用机制
#br#
帕金森病 (Parkinson's disease, PD)是第二常见的神经退行性疾病,α-突触蛋白(α-synuclein,α-syn)在多巴胺神经元(Dopaminergic, DA)内的异常聚集和DA神经元进行性丢失是其主要病理特征,然而目前其具体发病机制尚不明确[1]。α-syn作为帕金森病的致病蛋白,其降解主要依赖于自噬溶酶体途径及分子伴侣介导的自噬途径[2]。囊泡ATP酶(Vacuolar H+-ATPase, V-ATPase)是一种依赖于ATP的质子泵,通过调控细胞内外的H+浓度,维持各细胞器的正常生理功能,特别是溶酶体[3]。囊泡ATP酶也与自噬密切相关 [4]。ATP6V0A1是V-ATPase的V0区a1亚基,高丰度表达于神经元上[5]。在果蝇的阿尔茨海默病模型上,ATP6V0A1缺失或突变的神经元对病理蛋白神经毒性的易感性,但在帕金森病上仍无相关研究[6]。ATP6V0A1是否能调控自噬,能否通过自噬介导α-syn的降解,进而保护多巴胺神经元,减缓帕金森病进展,目前尚不清楚。
广州医科大学附属第一医院徐评议团队在发表于《中国神经再生研究(英文)》杂志(Neural
Regeneration Research)的研究发现,ATP6V0A1可明显降低α-syn在帕金森病模型上的异常聚集,减缓多巴胺神经元的丢失,改善帕金森病模型小鼠的运动能力。进一步机制研究证实,ATP6V0A1可通过调控溶酶体功能、调控mTORC通路及自噬溶酶体融合,降低α-syn的异常聚集而发挥保护效应。该文带来提示:ATP6V0A1在帕金森病上具有保护作用,未来或能成为帕金森病治疗的潜在靶点。第一作者为林钰婉、谭子欣、叶文峰。通讯作者为陈祥、徐评议、张文龙。
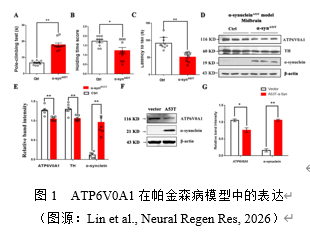
帕金森病发病原因复杂且不明确,环境毒素与基因易感性是其两大原因 [7]。实验团队前期发现ATP6V0A1 rs601999 位点突变可增加帕金森病患病风险[8]。为此,实验在α-synA53T基因鼠上观察ATP6V0A1蛋白水平,进一步评估其与帕金森病相关性。α-synA53T基因鼠上,小鼠的运动能力下降,表现为:α-synA53T基因鼠爬杆时间增加,悬挂评分及滚轴运动持续时间减少。α-synA53T基因小鼠中脑酪氨酸羟化酶(Tyrosine Hydroxylase, TH)和α-syn 表达下降,以上结果提示帕金森病模型建立成功(图1)。此外,在中脑α-synA53T小鼠及α-synA53T细胞稳转株上,ATP6V0A1表达减少(图1),这些结果表明ATP6V0A1在帕金森病动物及细胞模型上蛋白水平降低,提示其与帕金森病相关。
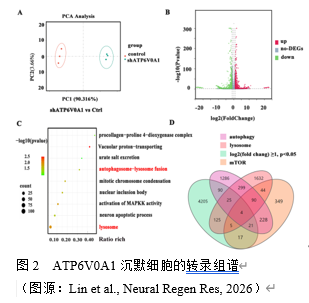
实验通过检测ATP6V0A1在α-synA53T动物及细胞模型的蛋白水平,初步证实了其与帕金森病相关,但具体机制尚不明确。为此,实验通过转染ATP6V0A1沉默慢病毒并对其进行RNA 测序分析,以评估其参与帕金森病的具体机制,结果提示:在主成分分析中,对照组和 ATP6V0A1 沉默组基因分集在两侧,对照组基因主要聚集在 PC1 的左侧,而ATP6V0A1沉默组则聚集在右侧,且两者区分度达到90.316%,表明两组之间存在明显的差异(图 2)。此外,筛选出表达变化至少2倍差异的基因,并通过火山图进行展示,红色为表达上调的差异基因,绿色则代表表达下降的差异基因。结果表明:差异基因总共有3050 个,其中 1333 个为上调基因,1717 个为下调基因(图 2)。进一步基因富集分析:大量差异基因富集于溶酶体与自噬上,其中140 个与自噬相关,159 个与溶酶体功能相关,47 个与 mTOR 通路相关,提示:ATP6V0A1在自噬及mTOR 通路发挥关键作用,或为其参与帕金森病的具体机制(图 2)。
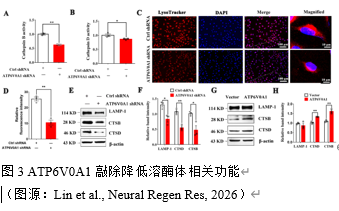
实验通过RNA测序分析发现ATP6V0A1与溶酶体功能密切相关。为此,为进一步探讨ATP6V0A1对溶酶体功能的影响,实验者采用组织蛋白酶B(Cathepsin
B, CTSB)和组织蛋白酶D (Cathepsin D, CTSD) 活性测定试剂盒分别测定CTSB及CTSD的活性。结果表明:ATP6V0A1沉默细胞株CTSB和CTSD活性下调 (图3)。实验者使用LysoTracker探针评估多巴胺能神经元酸性强度以明确ATP6V0A1对细胞pH稳态的影响,发现:ATP6V0A1沉默可降低溶酶体酸度;同样地,蛋白水平上,ATP6V0A1沉默可下调CTSB 和CTSD的蛋白水平,并降低溶酶体标志物LAMP-1的蛋白水平,而过表达ATP6V0A1可上调CTSB
和CTSD的蛋白水平(图3)。以上结果提示:ATP6V0A1参与调控溶酶体生理功能,包括溶酶体酶的活性及表达,参与调控溶酶体酸度。
自噬对于维持细胞内稳态、促进自噬体的形成、长寿命蛋白质或细胞器的吞噬以及自噬体与溶酶体的融合至关重要[9]。自噬是α-syn降解的重要途径,自噬紊乱可导致α-syn异常聚集,加重帕金森病进展。实验结合上述ATP6V0A1对溶酶体功能的影响及RNA 测序结果,进一步探索ATP6V0A1沉默对自噬通量的影响。实验发现ATP6V0A1沉默可阻断自噬流,表现为上调p62和LC3II的蛋白水平(图4)。此外,透射电镜显示ATP6V0A1沉默细胞株上的自噬小体数量增加。免疫荧光染色显示ATP6V0A1沉默组的LAMP-1表达显著降低而 LC3表达显著增加(图4)。
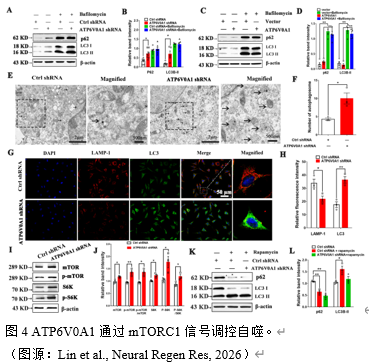
哺乳动物雷帕霉素复合物1靶点(mammalian target of rapamycin complex 1, mTORC1)信号通路是自噬的关键调控因子,参与调控自噬的各个阶段[10]。实验证明了ATP6V0A1沉默可阻断自噬流。但其是否通过mTORC1通路仍未可知。实验检测了空载组与ATP6V0A1沉默组mTORC1和S6激酶(S6K kinase,S6K)及其磷酸化蛋白表达水平,结果显示沉默组的p-mTORC1 / mTORC1和p-S6K/S6K比值较正常组升高,表明mTORC1-S6K信号被激活(图4)。然而,在100 nM雷帕霉素刺激24h后,两组之间p62或LC3II的蛋白水平无显著差异(图4),提示ATP6V0A1通过mTORC1信号调控自噬流。
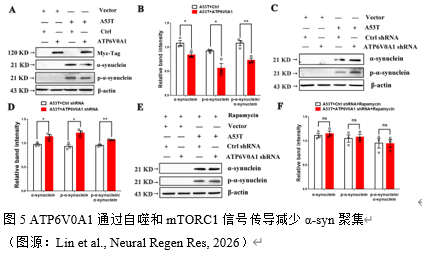
自噬-溶酶体途径是病理蛋白质降解的重要途径。实验通过上述实验初步证实了ATP6V0A1通过mTROC通路调控自噬,为进一步证实ATP6V0A1能否通过自噬影响α-syn的降解。实验通过双转染A53T-α-syn与ATP6V0A1沉默/过表达慢病毒,免疫蛋白印记检测α-syn及其磷酸化水平,发现:ATP6V0A1沉默可上调α-syn和p-α-syn蛋白水平并增加p-α-syn/α-syn比值(图5),而ATP6V0A1过表达下调α-syn 和p-α-syn蛋白水平并下降p-α-syn/α-syn比值(图5)。鉴于ATP6V0A1沉默可上调mTORC1信号而阻断自噬流,实验进一步探索mTORC1信号是否影响α-syn蛋白水平。结果显示:雷帕霉素刺激后,ATP6V0A1沉默组的p-α-syn和α-syn的蛋白水平及p-α-syn/α-syn比值无明显改变,提示ATP6V0A1通过影响mTORC1调控α-syn降解(图5)。
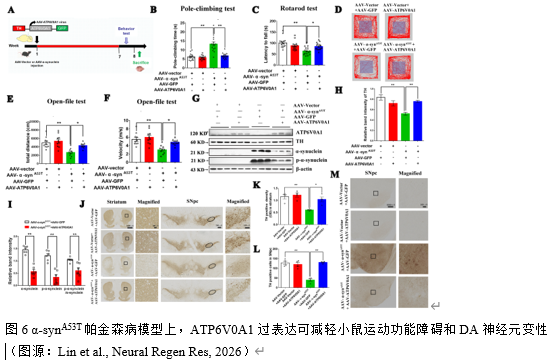
实验在帕金森病细胞模型上证实了ATP6V0A1可调控自噬介导α-syn的降解。为明确ATP6V0A1在帕金森病动物模型上的作用,实验通过在黑质区注射AAV-h-α-synA53T和AAV-ATP6V0A1腺相关病毒建立小鼠模型。6周后,行为学检测显示:ATP6V0A1可改善帕金森病小鼠的运动能力,表现为降低帕金森病小鼠爬杆时间,增加小鼠滚轴运动时间。矿场实验上,AAV-ATP6V0A1可增加帕金森病小鼠运动总距离和运动速度(图6)。分子水平上,ATP6V0A1下调帕金森病小鼠中脑组织α-syn、p-α-syn 蛋白水平和p-α-syn/α-syn比值,同时上调TH蛋白水平升高(图6),提示ATP6V0A1对DA神经元的保护效应。免疫组化叶证实了上述结果,ATP6V0A1可改善帕金森病小鼠黑质及纹状体TH的丢失,同时降低α-syn的阳性表达(图6)。这些结果表明,ATP6V0A1在黑质区DA神经元中过表达可减轻α-synA53T诱导的帕金森病表型。
综上所述,该研究结果表明ATP6V0A1调控自噬介导α-syn的降解,减缓DA神经元变性,改善帕金森病小鼠行为障碍。此项研究结果提示ATP6V0A1在帕金森病模型上的神经保护作用及其或能作为帕金森病治疗潜在靶点。#br#
然而,该研究仍存在一些局限性。首先,实验者未进行帕金森病患者的TP6V0A1全基因测序,未能明确R741Q或A512P位点与帕金森病风险的相关性,这些位点已被报道与脑发育相关的。再者,实验采用α-synA53T腺病毒构建帕金森病模型,但无法证实ATP6V0A1对α-synA53T转基因小鼠的保护效应。在机制上,实验聚焦于溶酶体自噬,但在动物水平上未能进行验证;因此,未来研究应基于原有的临床数据库上,扩大收集PD样本对ATP6V0A1进行全基因组测序,明确ATP6V0A1位点与帕金森病风险的相关性。此外,将构建双基因杂交小鼠,在体内水平上对溶酶体功能、溶酶体自噬和α-syn降解进行深入研究,充分证实ATP6V0A1在帕金森病的保护效应。#br#
#br#
#br#
参考文献#br#
1.
Guo M, Wang J, Zhao Y, et al. Microglial exosomes facilitate α-synuclein transmission in Parkinson's disease. Brain. 2020;143(5):1476-1497.#br#
2.
Xilouri M, Brekk OR, Stefanis L. α-Synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol. 2013;47(2):537-551.#br#
3.
Santos-Pereira C, Rodrigues LR, Côrte-Real M. Plasmalemmal V-ATPase as a potential biomarker for lactoferrin-based anticancer therapy. Biomolecules. 2022;12(1):119.#br#
4.
Satarker S, Bojja SL, Gurram PC, et al. Astrocytic glutamatergic transmission and its implications in neurodegenerative disorders. Cells. 2022;11(7):1139.#br#
5.
Schulz N, Dave MH, Stehberger PA, et al. Differential localization of vacuolar H+-ATPases containing a1, a2, a3, or a4 (ATP6V0A1-4) subunit isoforms along the nephron. Cell Physiol Biochem. 2007;20(1-4):109-120.#br#
6.
Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141(7):1146-1158.#br#
7.
Bogers JS, Bloem BR, Den Heijer JM. The etiology of Parkinson's disease: new perspectives from gene-environment interactions. J Parkinsons Dis. 2023;13(8):1281-1288.#br#
8.
Chen X, Xiao Y, Guo W, et al. Relationship between variants of 17 newly loci and Parkinson's disease in a Chinese population. Neurobiol Aging. 2019;73:230.e1-230.e4.#br#
9.
Corti O, Blomgren K, Poletti A, et al. Autophagy in neurodegeneration: New insights underpinning therapy for neurological diseases. J Neurochem. 2020;154(4):354-371.#br#
10.
Noda T. Regulation of autophagy through TORC1 and mTORC1. Biomolecules. 2017;7(3):52.#br#
#br#