中国神经再生研究(英文版) ›› 2026, Vol. 21 ›› Issue (4): 1628-1640.doi: 10.4103/NRR.NRR-D-24-00461
实验性青光眼Müller细胞内向整流钾通道Kir4.1和Kir4.1 Tyr9Asp突变过表达的神经保护作用
Overexpression of the inwardly rectifying potassium channel Kir4.1 or Kir4.1 Tyr9 Asp in Müller cells exerts neuroprotective effects in an experimental glaucoma model
Fang Li1 , Zhen Li1 , Shuying Li1 , Hong Zhou1 , Yunhui Guo1 , Yongchen Wang2 , Bo Lei2, 3, Yanying Miao1, *, Zhongfeng Wang1, *
- 1 State Key Laboratory of Medical Neurobiology and MOE Frontiers Center for Brain Science, Institute of Brain Science, Fudan University, Shanghai, China; 2 Institute of Neuroscience and Third Affiliated Hospital, Zhengzhou University, Zhengzhou, Henan Province, China; 3 Henan Provincial People’s Hospital, Henan Eye Institute, Henan Eye Hospital, People’s Hospital of Zhengzhou University, Zhengzhou University, Zhengzhou, Henan Province, China
摘要:
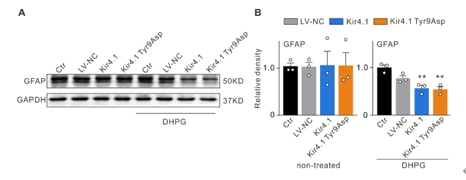
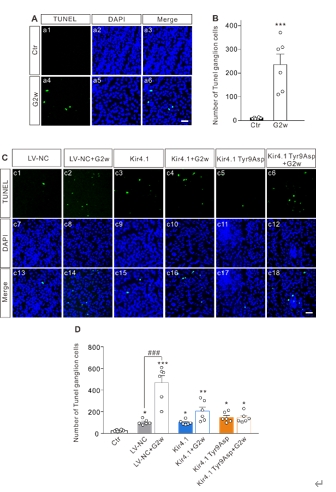
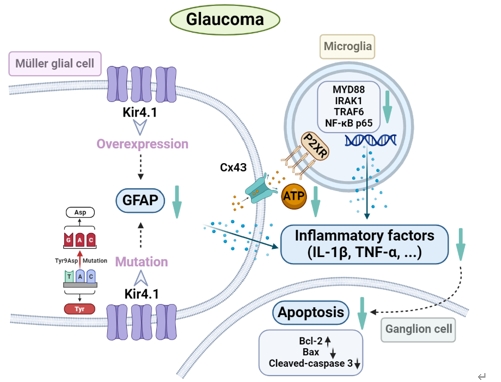
内向整流钾通道Kir4.1的表达和功能下调是诱导视网膜Müller细胞活化和神经胶质细胞相互作用的关键步骤,可参与青光眼视网膜神经节细胞的凋亡。因此调节Müller细胞中Kir4.1的表达可能是减轻青光眼视网膜神经节细胞损伤的潜在策略。此次实验中首先预测了Kir4.1蛋白中的7个磷酸化位点可发生突变,继而构建了含有不同突变位点的Kir4.1慢病毒表达载体,然后在mGluR I的激动剂DHPG处理以激活Müller胶质细胞的离体及在体模型中过表达Kir4.1和突变Kir4.1第9位酪氨酸,发现可抑制Müller胶质细胞的激活。接着在眼前房注射微磁珠建立的大鼠慢性高眼压动物模型中,发现Kir4.1过表达和Kir4.1 Tyr9Asp突变的Müller细胞中获得了类似的结果。过表达Kir4.1和Kir4.1 Tyr9Asp突变可抑制Müller胶质细胞激活,调控Bax/Bcl-2的平衡以及减少促炎因子白细胞介素1β和肿瘤坏死因子α的mRNA和蛋白质水平。进而在Müller胶质细胞和小胶质细胞共培养系统中研究了Müller胶质细胞Kir4.1过表达和Kir4.1 Tyr9Asp突变对促炎因子释放的调控作用,发现在共培养系统中,激活Müller细胞中ATP浓度和转运蛋白(小胶质细胞激活的标志物)水平升高,活化的Müler细胞诱导的小胶质细胞中炎症因子释放相关信号分子水平升高,上述变化可Kir4.1过表达和Kir4.1 Tyr9Asp突变所逆转。此外,Kir4.1过表达,而不是Kir4.1 Tyr9Asp突变,可减少活化的Müller细胞诱导的增殖和迁移小胶质细胞的数量。上述结果表明,在实验性青光眼视网膜中,Kir4.1 Tyr9可能是一个功能性调节位点。Kir4.1过表达和Kir4.1 Tyr9Asp突变减弱Müller细胞活化,减少ATP/P2X受体介导的神经胶质细胞相互作用,进而抑制小胶质细胞活化,降低促炎因子的合成和释放,从而减轻青光眼视网膜神经节细胞的凋亡。
https://orcid.org/0000-0002-2279-6885 (Zhongfeng Wang); https://orcid.org/0000-0003-3948-2943 (Yanying Miao)