中国神经再生研究(英文版) ›› 2024, Vol. 19 ›› Issue (4): 915-922.doi: 10.4103/1673-5374.382253
STAT3可改善截短tau蛋白所致认知功能障碍
STAT3 ameliorates truncated tau-induced cognitive deficits
Bingge Zhang1, #, Huali Wan2, #, Maimaitijiang Maierwufu1, Qian Liu1, Ting Li1, Ye He1, Xin Wang1, Gongping Liu1, Xiaoyue Hong3, *, Qiong Feng4, *
- 1Department of Pathophysiology, School of Basic Medicine and the Collaborative Innovation Center for Brain Science, Key Laboratory of Ministry of Education of China and Hubei Province for Neurological Disorders, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei Province, China; 2Department of Laboratory Medicine, Guangdong Provincial, People’s Hospital (Guangdong Academy of Medical Sciences), Southern Medical University, Guangzhou, Guangdong Province, China; 3Department of Laboratory Medicine, Zhongnan Hospital of Wuhan University, Hubei, Wuhan, Hubei Province, China; 4Department of Pathology, Wuhan Children’s Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei Province, China
摘要:
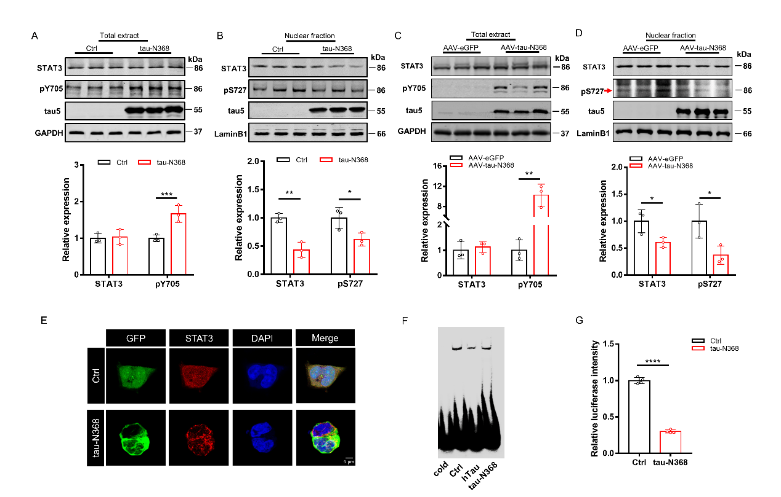
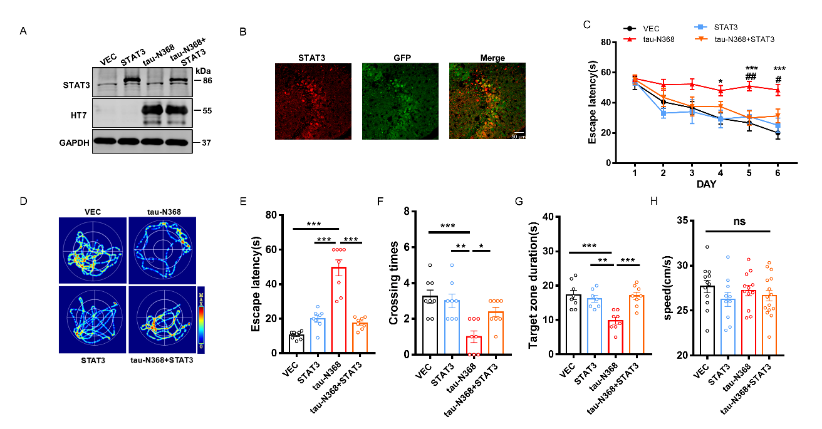
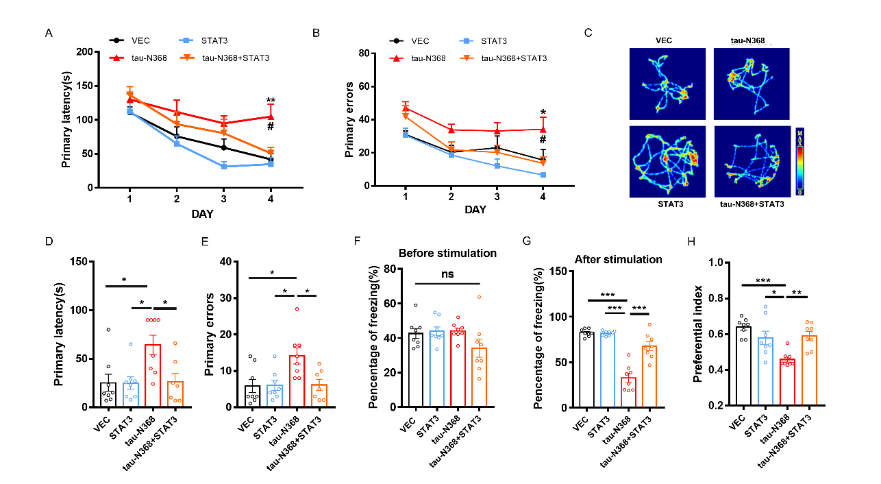
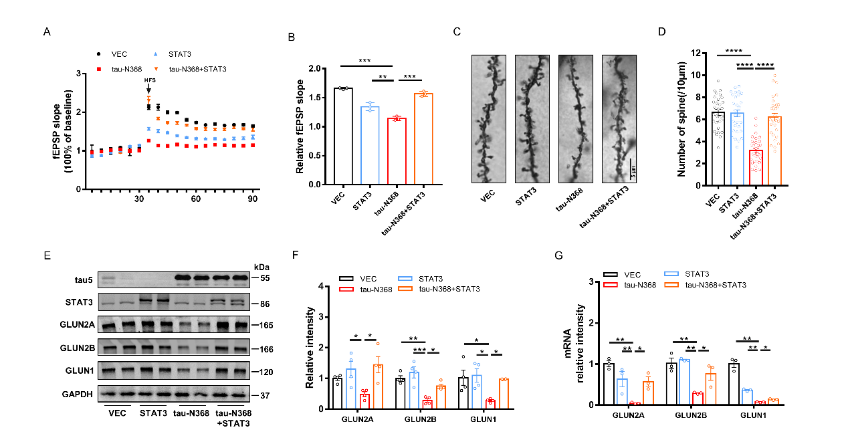
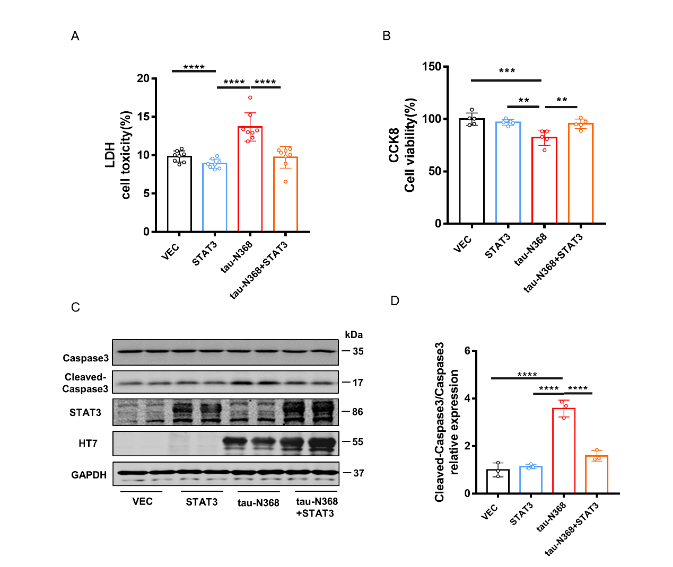
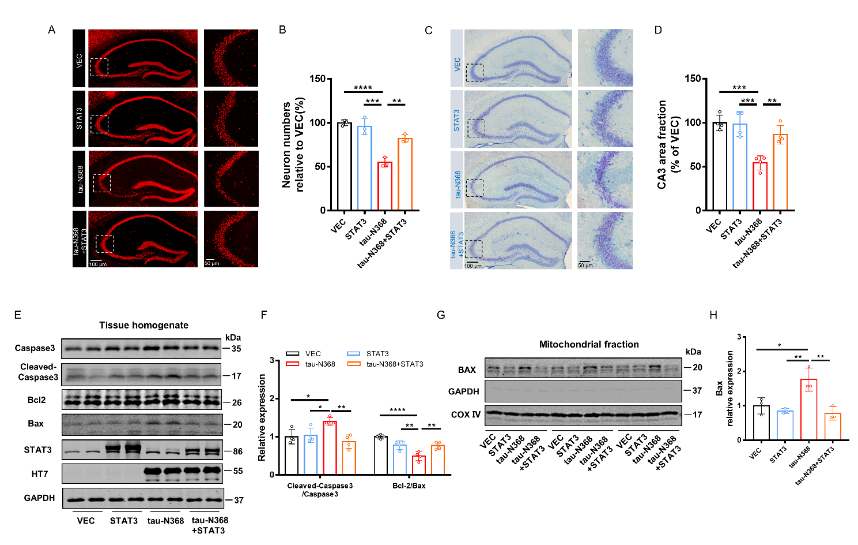
天冬酰胺内肽酶切割tau蛋白产生的tau N368片段,可能驱动阿尔茨海默病患者大脑中与突触功能障碍和记忆退化相关的病理变化,但是截短tau诱导认知障碍的分子机制仍有待探索。有证据表明,信号转导和转录激活因子3(STAT3)与调节突触可塑性、细胞凋亡和认知功能有关。此项实验通过萤光素酶报告基因分析、电泳迁移率偏移分析、蛋白质印迹分析以及免疫荧光分析发现,在HEK293细胞中人tau-N368的积累可通过抑制STAT3核转运来抑制STAT3活性。而过表达STAT3可改善tau-N368诱导的突触可塑性损伤,减少神经元的损失,从而改善tau-N368小鼠的认知障碍。同时在tau-N368小鼠中,可通过激活STAT3增加N-甲基-D-天冬氨酸受体蛋白水平,降低Bax蛋白水平,进而逆转突触损伤和神经元损失,从而缓解tau-N368引起的认知障碍。综上,STAT3对截短tau相关神经病理变化起着重要作用,这提示了一种tau-N368影响突触和记忆障碍的新机制,并且STAT3可作为治疗tau-N368诱导的tau蛋白病理的新分子靶标。
https://orcid.org/0009-0009-3690-411X (Xiaoyue Hong)